
Abstract
Monoclonal antibodies (mAbs) have emerged as a promising new drug class for the treatment of multiple myeloma (MM). Daratumumab (DARA), a CD38 mAb, has demonstrated safety, tolerability and activity in a range of clinical trials, both as monotherapy and in combination strategies for MM. The favorable efficacy results in heavily pretreated patients with advanced MM have provided the rationale for the investigation of DARA in a number of ongoing and future phase II and III trials. The general tolerability of mAbs has allowed for widespread investigation and use of DARA among a variety of MM patients, however their use requires special consideration. Infusion-related reactions (IRRs), interference with blood compatibility assays and response assessments are all unique factors related to the use of DARA. This review provides an update of the results from the DARA clinical trials conducted to date, its future plans for investigation, and practical management considerations for the use of DARA in daily practice.
Introduction
The survival of patients with multiple myeloma (MM) has markedly improved in the last decade, largely in part due to the introduction of novel treatment agents [van de Donk and Lokhorst, 2013]. However, despite substantial progress, patients with disease refractory to both immunomodulatory drugs (IMiDs) and proteasome inhibitors (PIs) have a median overall survival (OS) of only 9 months [Kumar et al. 2012]. The introduction of immunotherapy into the treatment landscape of MM, particularly monoclonal antibodies (mAbs), offers a promising new drug class with a different mechanism of action than has previously been available before in MM [Raje and Longo, 2015; Lonial et al. 2016a]. The mechanism of action, which includes a high specificity for antigens on the surface of the neoplastic cell and the capability to engage immune cells, presents a particularly exciting approach.
CD38 is a transmembrane glycoprotein that combines adhesion, receptor and enzymatic functions [Funaro and Malavasi, 1999; Mehta et al. 1996; Malavasi et al. 2008]. While the expression on normal lymphoid and myeloid cells is relatively low, CD38 is highly and uniformly expressed on myeloma cells. The mAbs against target antigens, such as CD38 expressed on MM cells, can induce tumor cell killing via a variety of mechanisms including complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity (ADCC), and antibody-dependent cellular phagocytosis [Ferris et al. 2010; Weiner et al. 2010; Overdijk et al. 2015].
CD38 has thus been identified as an attractive target for mAb therapy in MM and several anti-CD38 mAbs are currently being investigated in clinical trials. Daratumumab (DARA), an anti-CD38 mAb, was approved in November 2015 by the United States (US) Food and Drug Agency for the treatment of MM patients who have received at least three prior therapies including a PI and an IMiD, or who are double-refractory to these drugs. In this update on the use of DARA for the treatment of relapsed/refractory MM (RRMM), the preclinical and clinical development of this drug, both as monotherapy and in novel combinations, will be reviewed with the addition of practical clinical recommendations.
Preclinical activity
DARA, a human anti-CD38 immunoglobulin (Ig)G1k antibody was generated by immunization of transgenic mice possessing a human immunoglobulin gene with recombinant CD38 protein and NIH 353 cells until CD38-specific serum titer development. The antibodies generated were shown to have good binding to both Daudi, a B-lymphoblast cell line, and fresh MM cells [de Weers et al. 2011; Overdijk et al. 2015]. Preclinical studies also have shown synergy in inducing ADCC between both lenalidomide (LEN) or bortezomib and DARA. This synergistic effect was also seen in LEN/bortezomib-resistant MM cell lines and primary MM cells from bone marrow mononuclear cells derived from LEN- or bortezomib-refractory patients [Nijhof et al. 2015].
Clinical activity: daratumumab monotherapy
Two trials, the GEN501 and SIRIUS studies, initially evaluated the use of DARA monotherapy in early phase trials (Table 1). The GEN501 study was a phase I/II dose-escalation study of DARA, carried out in patients with RRMM who had received two or more prior lines of therapy and were ineligible for autologous stem cell transplant. The intent was to establish a safety profile, but to also evaluate pharmacokinetics and the maximum tolerated dose (MTD) as secondary objectives [Plesner et al. 2014; Lokhorst et al. 2015]. This 3 + 3 dose-escalation design with single-agent DARA treated 32 patients with gradually increasing doses ranging from 0.005 to 24 mg/kg without observation of a dose-limiting toxicity. Among 12 patients who received DARA 4–24 mg/kg, a partial response (PR) was seen in 4 patients and a minimal response (MR) was seen in 3 patients. Overall, DARA showed a favorable tolerability profile with serious adverse events (SAEs) occurring in 37% of patients, including: pyrexia and infections (9% each), bronchospasm (6%), anemia, thrombocytopenia, atrial fibrillation, abdominal pain, and hepatobiliary disorders (3% each). Infusion-related reactions (IRRs) were observed in 63% of patients, of which 6% were grade 3 or 4 (bronchospasm and hypersensitivity, 3% each). All patients recovered from their SAEs with treatment and the MTD was not reached. The predose (2 mg/kg) trough levels achieved were far below expected values, however doses of 4 mg/kg or greater resulted in adequate and sustained trough levels. Dose-dependent efficacy with greater decreases in paraprotein levels were seen in patients at higher doses. This trial was the first to demonstrate both substantial activity and good tolerability of DARA in patients with advanced disease and limited treatment options, and led to additional trials to investigate this promising drug.
Trials of daratumumab performed to date.

ASCT, autologous stem cell transplantation; CR, complete remission; DARA, daratumumab; DEX, dexamethasone; DOR, duration of response; DVD, DARA, bortezomib, and DEX; IMiD, immunomodulatory drugs; LEN, lenalidomide; MM, multiple myeloma; MR, NDMM, newly diagnosed multiple myeloma; NR, not reached; ORR, overall response rate; OS, overall survival; PI, proteasome inhibitors; PFS, progression-free survival; POM, pomalidomide; PR, partial response; RRMM, relapsed/refractory multiple myeloma; VGPR, very good partial response; sCR, stringent complete remission; VD, bortezomib and DEX; VMP, bortezomib, melphalan, prednisone; VTD, bortezomib, thalidomide, dexamethasone.
Lonial and colleagues published the results of a second DARA monotherapy study involving 106 patients, the MMY2002 SIRIUS trial [Lonial et al. 2016b]. Patients in this trial were heavily pretreated and had highly refractory disease, with a median of five prior lines of therapy. A total of 80% had undergone prior autologous stem cell transplantation and 95% had disease refractory to a PI and an IMiD. In addition, 63% of patients were refractory to pomalidomide (POM), 48% were refractory to carfilzomib, 66% were refractory to three of four therapies (bortezomib, LEN, carfilzomib, POM) and 31% were refractory to all four agents. An initial evaluation randomized patients 1:1 to receive either 8 mg/kg every 4 weeks (n = 18), or 16 mg/kg every week for 8 weeks, then every 2 weeks for 16 weeks, and continuing on with infusions every 4 weeks (n = 16). A response evaluation recommended 16 mg/kg as the recommended dose for further study and an additional 90 patients were enrolled at this dose level and schedule. For all 106 patients treated at 16 mg/kg, the overall response rate (ORR), (partial response or better) was 29.2% with 3 stringent complete remissions (sCRs), 10 VGPRs (serum and urine M-protein detectable by immunofixation but not on electrophoresis or ⩾90% reduction in serum M-protein plus urine M-protein level <100 mg/24 h) and 18 PRs, with a median time to response of 1 month and a median duration of response of 7.4 months [Lonial et al. 2016b]. These responses were seen across all subgroups evaluated, including: age ⩾ 75 years; more than three prior lines of therapy; and disease refractory to PIs and IMiDs. With a median follow up of 9.3 months, the median progression-free survival (PFS) was 3.7 months and the 1-year survival rate was 64.8%. An updated analysis demonstrated a median OS was 17.5 months. Treatment-related SAEs were noted in 30% of patients and grade 3/4 adverse events (AEs) were noted in 23% of patients. There were no discontinuations of drug due to AEs. The most frequent AEs of any grade were fatigue (40%) and anemia (33%). IRRs, seen in 42% of patients, occurred predominantly during the first infusion and were commonly grade 1 or 2 (5% grade 3, no grade 4), and were all manageable. Further, only 6% of patients had an IRR beyond the first infusion. The most common IRRs included nasal congestion (12%), throat irritation (7%), cough, dyspnea, chills and vomiting (6%). No patient discontinued treatment due to IRRs.
A combined analysis of the two monotherapy studies (GEN501, SIRIUS) was performed and included 148 patients who had received DARA 16 mg/kg (the recommended dose) [Usmani et al. 2016]. In this combined analysis 59 (39.9%) and 28 (18.9%) of 148 patients treated with DARA 16 mg/kg demonstrated ⩾50% and ⩾90% reductions in paraprotein from baseline, respectively. The median time to response in patients with PR or better was 0.95 months, and the ORR was 31.1% [95% confidence interval (CI), 23.7–39.2%]. The median duration of response was 7.6 months, and responses deepened with continued DARA treatment in 14 patients across the two studies. The median OS for the group was 20.1 months, with a 1-year OS rate of 69%. These results further demonstrate the significant single-agent activity of DARA in patients with very advanced-stage disease who are refractory to current treatment options, including the standard PIs and IMiDs, and when the typical survival is only 8–9 months [Kumar et al. 2012].
In order to evaluate the effects of DARA on immune cell populations and adaptive immune response pathways, peripheral blood and bone marrow samples were taken from 148 patients in the GEN501 and SIRIUS studies. In this population of heavily pretreated, relapsed and refractory patients who would otherwise not be expected to have strong immune responses, robust T-cell increases, increased CD8+:CD4+ ratios, increased antiviral T-cell responses, and increased T-cell clonality were all observed after DARA treatment. Importantly, improved clinical responses were associated with significant increases in these specific parameters. These data suggest a potentially important immunomodulatory role of DARA that may contribute to its efficacy [Krejcik et al. 2015].
Clinical activity: novel daratumumab combinations
DARA has been evaluated in combination with standard MM regimens (Table 2). An initial phase I/II study was performed on 45 patients with RRMM following a median of two prior therapies treated with combination DARA, LEN and dexamethasone (DEX) [Plesner et al. 2014]. DARA was dose-escalated from 2 to 16 mg/kg in part one of the study (n = 13), and then administered at 16 mg/kg in the expansion cohort (n = 32). LEN was administered at 25 mg on days 1–21, and DEX was administered at 40 mg weekly. With a median duration of follow up of 12.9 months for part one, the overall best response was 100% (31% CR, 46% VGPR, 23% PR). In the planned part two of the study, 32 LEN-sensitive patients with at least one prior line of therapy were treated at the 16 mg/kg dose of DARA + LEN/DEX. With a median follow up of 15.6 months, the ORR was 81% with 19% PR, 28% VGPR, 9% CR, and 25% sCR [Plesner et al. 2015]. The clinical benefit rate (⩾MR) was 88%, and 91% of patients were progression-free at 12 months. The median duration of response was not reached as 26 (93%) of 28 responders had not progressed or relapsed at the time of the analysis. Responses were found to deepen over time, with a time to first response of 1 month and time to best response of 5.1 months. The 18-month PFS and OS rates were 72 and 90%, respectively. The most frequent treatment-related AEs were neutropenia (84%), cough (50%), diarrhea (44%) and muscle spasms (44%), and the most frequent grade 3 or higher AEs were neutropenia (78%), thrombocytopenia (13%) and anemia (13%). A total of 16 patients had SAEs, 9 of which were due to infection. IRRs were seen in 19 (56%) patients, and were grade 2 or lower in the majority of patients. IRRs occurred during the first infusion with three patients experiencing IRRs in the second or subsequent infusions. In two patients, grade 3 IRRs were noted (laryngeal edema and hypertension), but no grade 4 IRRs were reported.
Upcoming and planned daratumumab trials.
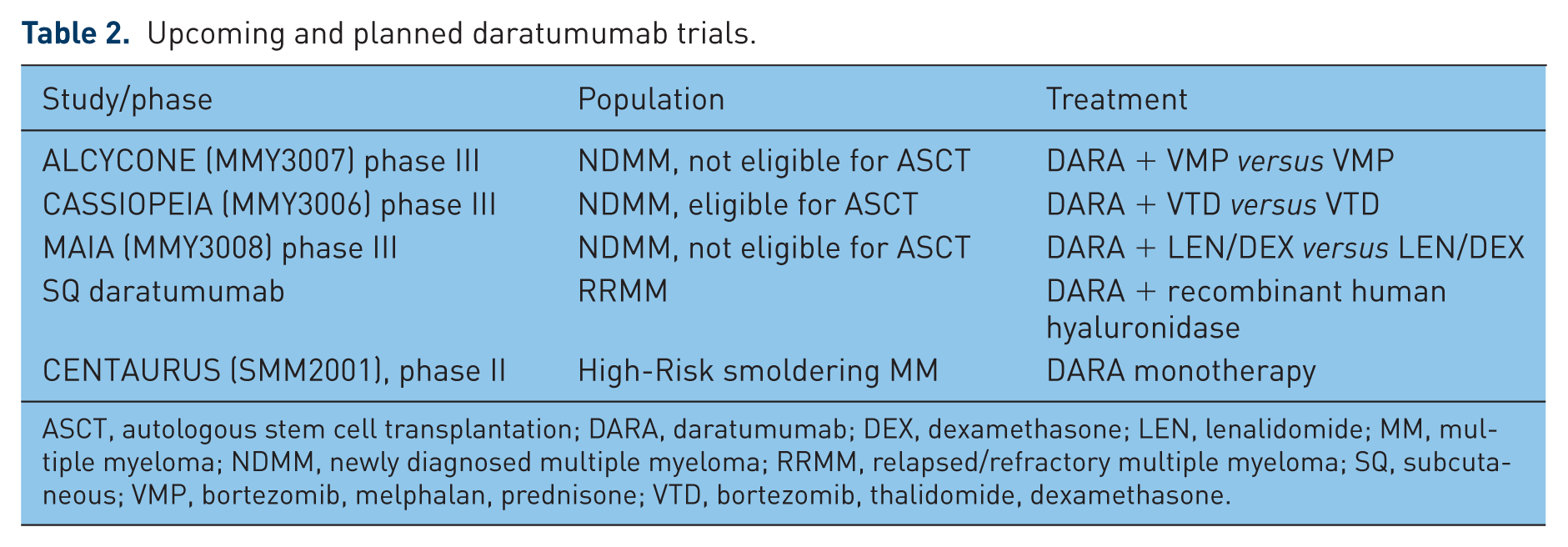
ASCT, autologous stem cell transplantation; DARA, daratumumab; DEX, dexamethasone; LEN, lenalidomide; MM, multiple myeloma; NDMM, newly diagnosed multiple myeloma; RRMM, relapsed/refractory multiple myeloma; SQ, subcutaneous; VMP, bortezomib, melphalan, prednisone; VTD, bortezomib, thalidomide, dexamethasone.
Additional combination options including DARA have also been evaluated in an open-label, four-arm, multicenter, phase Ib study [Mateos et al. 2016; Moreau et al. 2014; Chari et al. 2015]. In this trial, 25 patients with newly-diagnosed multiple myeloma (NDMM), regardless of transplant eligibility, were treated with DARA in combination with one of three arms: bortezomib and DEX (VD, n = 6), bortezomib, melphalan and prednisone (VMP, n = 8), or with bortezomib, thalidomide and DEX (VTD, n = 11). A fourth arm included an additional 24 patients with RRMM who were treated with the combination of pomalidomide and DEX (POM-D) with DARA. The ORR in 35 evaluable patients was 100% in the newly-diagnosed group (DARA + VD: 3 PRs, 3 VGPRs; DARA + VTD: 7 PRs, 2 VGPRs, 1 CR; DARA + VMP: 4 PRs, 4 VGPRs), with a median follow up of 6.4, 5.5, and 8.9 months for DARA + VD, DARA + VTD, and DARA + VMP, respectively [Mateos et al. 2016]. An updated efficacy analysis from Chari and colleagues at the 2015 American Society of Hematology annual meeting included a total of 77 RRMM patients in the DARA and POM-D arm with a median number of prior therapies of 3.5. Of the cohort, 65% were refractory to both PI and IMiD. The ORR was 58.5% with 3 sCRs, 1 CR, 12 VGPRs, and 15 PRs. After a median follow up of 4.9 months, 4/31 (13%) responders had progressed. Importantly, the combination of DARA and POM-D produced remarkable efficacy among 40 evaluable double-refractory patients, with a 57.5% ORR [Chari et al. 2015]. In the DARA and POM-D arm, IRRs were seen in 61% of patients, which most commonly included chills, cough and dyspnea, otherwise no significant additional toxicity was noted with the addition of DARA to the POM-D combination. Some of the most common AEs of any grade were neutropenia, anemia, fatigue, cough, nausea, dyspnea, and diarrhea. Grade 3 or 4 AEs occurring in 10% of patients were all hematologic toxicities [neutropenia (50.6%), anemia (20.8%), leukopenia (15.6%) and thrombocytopenia (10.4%)]. Importantly, DARA had no negative impact on stem cell mobilization.
The remarkable efficacy of DARA in combination with both LEN/DEX and POM-D in RRMM and in combination with other backbone regimens in NDMM has provided the rationale for the initiation of phase III trials. The MMY3003 POLLUX study, an open-label, randomized phase III study of DARA in combination with LEN and DEX versus LEN and DEX was performed in 529 patients with RRMM who had received a median of one prior line of therapy [Dimopoulos et al. 2016]. Patients were treated in both arms until progressive disease. The primary endpoint of the study was PFS with a preplanned interim analysis demonstrating a 63% reduction in risk of disease progression in those who received the combination of DARA, LEN and DEX compared with those who did not receive DARA [hazard ratio (HR) = 0.37; 95% CI, 0.27–0.52; p < 0.001]. The median PFS for the combination arm has not been reached, compared with an estimated PFS of 18.4 months of patients who received LEN and DEX alone.
The MMY3004 CASTOR study, a phase III randomized controlled study of DARA, bortezomib, and DEX (DVD) versus bortezomib and DEX (VD) in patients with RRMM, was performed in 498 patients [Palumbo et al. 2016]. Patients who had received at least one prior line of therapy were randomized to receive eight cycles of bortezomib (V) and DEX (D) (V: 1.3 mg/m2 subcutaneously on days 1, 4, 8, 11; D: 20 mg orally on days 1, 2, 4, 5, 8, 9, 11, 12) with or without DARA (16 mg/kg intravenously weekly in cycles 1–3, day 1 of cycles 4–8, then every 4 weeks). Patients randomized to the investigational arm received DARA until progression of disease, whereas the control arm completed therapy after eight cycles of VD and were subsequently monitored. Included patients had received a median of two prior lines of therapy (range, 1–10). Across the two treatment groups, 61.2% of the patients had undergone autologous stem cell transplantation, 65.5% had received previous treatment with bortezomib, 75.7% had received IMiDs, 48.4% had received both PIs and IMiDs, 32.3% had disease that was refractory to their last line of therapy, and 32.9% had disease that was refractory to IMiDs. With a median follow up of 7.4 months, the addition of DARA significantly improved the median PFS (61% reduction in risk of progression) and time to progression for DVD versus VD. The addition of DARA significantly increased the ORR to 83% versus 63% (p < 0.0001), and doubled rates of ⩾VGPR (59% versus 29%, p < 0.0001), and ⩾CR (19% versus 9%, p = 0.0012) for DVD versus VD respectively. The median duration of response was not reached in the DVD arm versus 7.9 months with VD. The most common AEs for DVD/VD were thrombocytopenia (59%/44%), peripheral sensory neuropathy (47%/38%), diarrhea (32%/22%) and anemia (26%/31%). The most common grade 3/4 AEs (>10%) were thrombocytopenia (45%/33%), anemia (14%/16%), neutropenia (13%/4%). Treatment was discontinued in 7% and 9% of patients due to treatment-related AEs in DVD and VD, respectively. IRRs due to DARA occurred in 45% of patients, and mostly occurred during the first infusion, with only 9% grade 3 reactions, and no grade 4 reactions.
Future clinical trials with daratumumab combinations
The early efficacy seen with the addition of DARA in NDMM patients has led to the initiation of the randomized, open-label multicenter, phase III trial ALCYCONE that will compare VMP and DARA-VMP in patients with newly diagnosed, previously untreated MM who are ineligible for high-dose therapy with stem cell transplantation. Another randomized, phase III trial, CASSOPEIA, will evaluate previously untreated NDMM, transplant-eligible patients with VTD, with or without DARA, as induction therapy prior to transplant followed by VTD with or without DARA as consolidation therapy. Patients will then be re-randomized to DARA maintenance therapy versus observation. Finally, a US multicenter, randomized, open-label, active-controlled, phase II study is planned in NDMM patients eligible for high-dose chemotherapy and autologous stem cell transplant. Patients will be randomized to receive DARA, in combination with LEN/bortezomib/ DEX (RVD) or RVD alone. All patients will undergo four 21-day induction treatment cycles followed by stem cell mobilization, high-dose chemotherapy and autologous stem cell transplantation, followed by two 21-day consolidation treatment cycles. All planned trials have PFS as the primary endpoint.
Practical considerations
Optimal schedule, infusion guidelines, and IRR management
Based on the available clinical trials, the optimal dose of DARA single-agent has been established at 16 mg/kg as an intravenous infusion, administered weekly during the first 8 weeks, every 2 weeks for the following 16 weeks, and every 4 weeks thereafter. The optimal duration of treatment is unknown, however based on currently ongoing trials, DARA is being administered until disease progression or unacceptable toxicity, as no long-term toxicity has been observed with DARA.
The infusion solution is prepared as a 1000 ml dilution of DARA in sterile 0.9% NaCl on the first planned infusion day, while subsequent solutions are prepared in 500 ml 0.9% NaCl. The drug is administered as an intravenous infusion, with each patient’s dose calculated based on the patient’s weight rounded to the nearest kilogram. It is recommended that the first infusion of DARA, diluted in 1000 ml NaCl, should be administered at an initial rate of 50 ml/h in the first hour, with subsequent increases at the rate of 50 ml/h every hour up to a maximum infusion rate of 200 ml/h [Lokhorst et al. 2015]. The same infusion rate should be used for the second infusion (dilution volume 500 ml), but subsequent infusions can be started at an initial rate of 100 ml/h and increased 50 ml/h every hour up to a maximum infusion rate of 200 ml/h. An increase in infusion rate should occur only if prior infusions were well tolerated by each individual patient. Careful monitoring for IRRs is of utmost importance where the infusion should be immediately paused for the emergence of symptoms, the symptoms treated accordingly, and the infusion resumed at a lower rate than when the symptoms occurred. Infusion duration for the first administration may last approximately 8 h, with subsequent infusions lasting approximately 4 h, assuming no IRRs.
The subcutaneous formulation of therapeutic antibodies with recombinant human hyaluronidase (rHuPh20) has been approved in Europe, and a current phase Ib study is evaluating the delivery of subcutaneous DARA in patients with symptomatic RRMM who have received at least two prior therapies in the United States. Varied dose cohorts will be established and both pharmacokinetic (PK) and safety data will be determined to obtain a recommended part two dose for further evaluation. Recruitment for this trial is ongoing [Nahi et al. 2016].
IRRs seen with mAbs are generally mild and only a small fraction of patients develop severe reactions [Chung, 2008; Guan et al. 2015]. The highest risk of reaction is during the first or second exposure to the mAb, with the risk declining with subsequent infusions. With DARA, IRRs were seen in 42–71% of patients, and occurred most commonly during the first infusion. IRRs were grade 1/2 in almost all cases, there were no grade 4 IRRs, and discontinuations due to IRRs were rare [Lokhorst et al. 2015; Lonial et al. 2016b]. Common IRRs with DARA include nasal congestion, throat irritation, cough, dyspnea, chills and vomiting. It is important to know that the occurrence of IRRs can be influenced by the infusion rate. In order to prevent IRRs with DARA, premedications given 1 h prior to DARA administration should include: intravenous corticosteroid (methylprednisolone 100 mg or an equivalent long-acting corticosteroid for the first two infusions, and 60 mg thereafter, in the absence of IRRs in the first two infusions), oral antipyretics (paracetamol, 650–1000 mg), and an oral or intravenous antihistamine (diphenhydramine, 25–50 mg or equivalent). If IRRs occur despite the premedication regimen, it is essential that the infusion be paused immediately, even if only mild symptoms are detectable. The infusion should be held until the appropriate symptom management is initiated and symptoms have resolved. DARA can be restarted at a lower infusion rate following the resolution of symptoms [Voorhees et al. 2015] (Table 3). Oral corticosteroids on the first and second days following DARA infusions (methylprednisolone, 20 mg; or equivalent) can help to prevent the occurrence of delayed IRRs. Patients with known obstructive pulmonary disease with a forced expiratory volume in 1 second (FEV1) < 50%, or with moderate or severe persistent asthma within the past 2 years, should be carefully considered as candidates for DARA due to the concern for bronchospasm, and any patient with a history of obstructive pulmonary disease should be considered for short- and long-term bronchodilators and inhaled corticosteroids.
Recommendations for the management of IRRs.

IRR, infusion-related reaction.
Management of daratumumab therapy
Blood typing
DARA does not interfere with ABO/RhD typing, however early-phase studies have documented an interference with routine laboratory tests used in blood transfusion medicine. In patients receiving DARA, the indirect antiglobulin test (IAT; Coombs test), used for the detection of irregular blood group antibodies, was found to be falsely positive [Oostendorp et al. 2015; Chapuy et al. 2015]. CD38 found on erythrocytes results in agglutination of the DARA to red blood cells, masking the detection of antibodies to minor antigens in the patient’s serum, leading to the false positive IAT result that is durable for up to 5 months. This can create a challenging scenario for transfusion medicine departments when conducting reliable blood compatibility testing. Strategies to overcome the interference of CD38 mAbs with IATs have been developed, including the denaturation of cell surface CD38 with the reducing agent dithiothreitol (DTT) and the addition of an excess of soluble CD38 or neutralizing anti-idiotype antibodies. Using these methods, irregular antibody screening and identification could be restored [Chapuy et al. 2015; Huber et al. 2011; Oostendorp et al. 2015]. In order to provide safe packed red blood cells (RBCs) for transfusion to patients receiving DARA, units should be ABO/RhD compatible, phenotypically or genotypically-matched units, and provide ABO/RhD compatible, K-negative units after ruling out or identifying allo-antibodies using DTT-treated reagent RBCs. In an emergency, uncrossmatched, ABO/RhD compatible RBC units should be administered, as per local transfusion medicine department practice.
Response assessment
Updated guidelines of the International Myeloma Working Group response criteria require the absence of an M-protein on serum protein electrophoresis (SPEP) and serum immunofixation electrophoresis (SIFE) as a requirement for a CR [Palumbo et al. 2014].
Therapeutic mAbs such as chimeric human-mouse Igs (rituximab) and human mAbs (ofatumumab) have previously demonstrated an interference with SPEP, leading to false positive results [Genzen et al. 2011; McCudden et al. 2010]. The mAbs, as Igs, can be identified on SPEP and SIFE, preventing the distinction between the therapeutic antibody and the patient’s clonal Ig, thereby confounding the response assessment. To help distinguish DARA from endogenous M-protein, a DARA-specific immunofixation electrophoresis reflex assay was developed to confirm suspected DARA interference and to allow separation of DARA bands from residual endogenous M-protein [McCudden et al. 2016]. This assay has been developed and validated for DARA and involves the addition of an anti-idiotype mAb to patient samples which binds to DARA [McCudden et al. 2015].
Daratumumab in specific populations
Liver dysfunction
No dose modifications are necessary for patients with mild hepatic impairment based on population pharmacokinetic analysis. No data are available for moderate or severe hepatic impairment (accessed 19 July 2016).
Renal dysfunction
DARA is not metabolized by the kidney; such that renal failure is not a contraindication for treatment. The GEN501 and SIRIUS trials each included patients with mild-to-moderate renal failure, creatinine clearance 30–60 ml/min and the ORR in these patients was 26.2%. [Lonial et al. 2016b]. No data are available to provide guidance on patients with severe renal impairment.
Advanced age
The GEN501 was administered to 16 patients aged 65–74 years, 56% of whom responded [Lokhorst et al. 2015], while none of the 4 patients over age 75 responded. In the SIRIUS trial, 36 patients were aged 65–74 years, and 12 patients were 75 years or older. The ORR in these subgroups of patients was 25% and 33.3%, respectively, suggesting that the efficacy of DARA is equivalent in all age groups.
Future directions
As discussed above, the future of DARA includes its evaluation in a number of clinical trials for MM (Table 2). Both the CASTOR and POLLUX studies will have updated analyses of their primary endpoint measure of PFS. The ALCYONE trial is planned to enroll 700 patients to receive DARA + VMP versus VMP alone in a randomized phase III trial. The IFM/HOVON cooperative trial evaluating DARA in the transplant setting (CASSIOPEIA) will randomize 1000 patients to receive VTD ± DARA as induction and consolidation, followed by re-randomization to DARA maintenance versus observation. Similarly, in the US, the upcoming MMY3004 study of RVD ± DARA will evaluate patients with NDMM in the transplant-eligible setting. Future trials will also investigate DARA in high-risk smoldering myeloma in the CENTAURUS trial, in addition to its use in other hematological malignancies.
Conclusion
The introduction of CD38 mAbs to the treatment landscape of MM has proven to be transformative. Given its unique mechanism of action as well as favorable tolerability, DARA offers tremendous promise in further improving outcomes in patients with MM, both in the RRMM and NDMM populations.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
Dr Costello serves on a speaker’s bureau for Janssen (Titusville, NJ, USA).