
Abstract
Hemophilia A, characterized by impaired or absent expression of factor VIII, has long been managed via direct factor replacement. Functionally, factor VIII acts as a cofactor for factor IXa and allows activation of factor X, which, in combination with factor V, generates thrombin. Bispecific antibodies such as emicizumab are recombinant, monoclonal antibodies capable of recognizing and binding to two distinct antigenic targets simultaneously; emicizumab binds factors IXa and X, resulting in spatial approximation and activation of factor X, thereby mimicking the actions of factor VIII. Critically, the presence of antifactor VIII antibodies, for example, inhibitors, impacts neither the mechanism nor the efficacy by which emicizumab functions. The results and interim analyses of the emicizumab clinical trials, HAVEN 1, 2, 3, and 4, are additionally reviewed and discussed.
Keywords
Introduction: providing a context for emicizumab
‘For it was taught: If she circumcised her first child and he died, and a second one who also died, she must not circumcise her third child…It once happened with four sisters that when the first had circumcised her child he died; when the second circumcised her child he also died, and when the third circumcised her child he also died. The fourth was told “You must not circumcise this child.”’
Hemophilia: past and present
The first modern medical description of hemophilia was provided by John Conrad Otto of Philadelphia, in 1803. In his paper he described the cardinal features of the condition, including its heritable nature, male preponderance, and bleeding tendency. 1 The first description of any kind is found in a far earlier text, the Talmud, and dates from the 2nd century AD.
As a distinct disease entity, hemophilia has evidently been long recognized. Its pathophysiological etiology has, at least within the context of modern medicine, also been understood for a relatively long time. Patek and Taylor showed that, via the addition of a blood-derived substance they referred to as ‘antihemophilic globulin’ to hemophiliac plasma, coagulation defects could be corrected. 2 The various clotting factors, including VIII, were discovered and named during the late 1950s and early 1960s, and the coagulation cascade first definitively described in 1964.3,4
With a firm pathophysiologic basis established, rational treatment became possible. Prior to the 1970s, and Judith Pool’s 1964 discovery of the large quantity of factor VIII (as well fibrinogen, factor XIII, von Willebrand factor, and fibronectin) contained in the residual precipitate formed from thawing plasma, 5 whole blood and fresh plasma were the only available treatment options. However, as the concentration of factor VIII contained therein is wholly insufficient to halt severe hemorrhage, the vast majority of those with severe hemophilia died in childhood. 6
Hemophilia treatment in the modern era
The development of lyophilized factor concentrate formulations in the 1970s was a harbinger of modern hemophilia treatment. Derived from the pooled plasma of thousands of blood donors, lyophilized factor concentrate was practical for home use. Available as a reconstitutable powder, patients and their caregivers were for the first time able to administer therapy outside the hospital setting, with resultant improved control over hemorrhages and their sequelae, and a subsequent drop in mortality. 7 It was, however, during this period that factor VIII inhibitors started becoming a major concern. Prothrombin complex concentrates were used initially on an ad hoc basis to address this problem, and later in a systematic manner once their safety and efficacy was established by Jeanne Lusher and others.8 –10 At that time, the massive progress in hemophilia treatment was heralded as one of the decade’s major medical triumphs.
The optimism of that era was shattered in the early 1980s by the onset of the human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome crisis. Over two thirds of those with severe hemophilia were infected and subsequently died after acquiring the virus from contaminated factor concentrates; almost all treated patients in that era also acquired the hepatitis C virus, though the long-term sequelae of cirrhosis and hepatocellular carcinoma were not well understood at the time. 7 These events, and the subsequent devastation they wrought within the hemophilia community, both underscored the need for and drove the development of improved viral inactivation and screening methods. Moreover, the sequencing and cloning of the factor VIII gene in 198411 –13 facilitated the manufacture of recombinant factor concentrates, thereby providing a viable therapeutic alternative to human-derived blood products for the first time; 1989 saw the first reported use of recombinant factor VIII in hemophilia A. 14 Since that time, rapid pharmacological research and near-continuous development of new forms of recombinant factor have driven the field forward at a rapid pace; the most recent generation of products are totally recombinant, without the presence or addition of animal or human proteins at any point in the manufacturing process. Human plasma-derived factor concentrates remain available and are now exceptionally safe; the last known transmission of HIV via plasma-derived factor concentrate occurred over 30 years ago. 15 Though heavily utilized elsewhere in the world, they remain less popular in the United States.
The individual patient’s response to factor replacement dosing is highly variable and must be individualized. In those without hemophilia, factor VIII is produced at a rate of approximately 3 units/kg/h. 16 Factor VIII circulates, bound to von Willebrand factor, which shields it from degradation; plasma von Willebrand concentration therefore influences clearance rate. Individualized pharmacokinetic studies are necessary to optimize dosing; this requires administration of a dose of factor replacement, with levels obtained prior to and at set timepoints following dosing. 17 Population pharmacokinetic modeling (e.g. Bayesian analysis) has allowed the development of predictive models that enable a reduced number of timepoints (e.g. at 4, 24, and 48 h) to be utilized to develop an individualized dose-response curve, thereby improving economy and feasibility of pharmacokinetic-guided dosing. 18
The present state of affairs is such that a child born with hemophilia A may be expected to have a normal life expectancy. 19 Frequent prophylactic infusions, typically three to four times per week, are required to maintain an adequate trough level to prevent hemorrhagic complications due to the comparatively short half-life of factor VIII preparations (approximately 8–12 h, though with significant interpatient variability, especially in children). 20 Efforts to extend the half-life of recombinant factor have been met with modest success, and recent recombinant products have achieved half-lives of approximately 19 h; 1.4–1.5 times longer than standard factor VIII preparations. 20
Factor VIII inhibitors in hemophilia A
Assuming an adequate frequency of dosing, factor VIII replacement is a highly effective, safe, and feasible treatment modality. However, there remain major challenges, the most significant of which is the tendency of patients to develop inhibitors. An inhibitor is a high affinity, polyclonal immunoglobulin G (IgG) against factor VIII. 21 Factor VIII comprises six domains: three A domains (A1–3), one B domain, and two C domains (C1, C2). Due to its structure, antibody binding to the A2, A3, or C2 domain physically occludes factor VIII’s functional epitopes, including binding sites for von Willebrand factor and factor IX, thereby impeding functional involvement in the coagulation cascade. 21 Regardless of formulation or source, all forms of factor replacement are immunogenic, and an inhibitor may form against any of them. Thus, managing those patients that do develop inhibitors is a key ongoing focus in the field.
Broadly speaking, inhibitors are classified based on their kinetics. Type 1 inhibitors demonstrate second-order (linear) kinetics and inhibit factor VIII in a dose-dependent fashion up to and including complete inactivation, while type 2 inhibitors are more variable and demonstrate nonlinear kinetics and incomplete factor VIII inactivation. 21
Measurement of inhibitors
The Bethesda assay, and the Nijmegen-modified Bethesda assay which succeeded it, are the primary means of detection and quantification of inhibitors. In both tests, serial dilutions of a patient’s plasma and an equal volume of nonfactor deficient plasma are performed, and the residual factor activity is measured, a positive test being one which identifies a significant decrease in factor activity, signifying the presence of an inhibitor. The residual factor and the dilution are plotted, and the inhibitor titer obtained via linear regression, one Bethesda unit (BU) being the amount of an inhibitor which inhibits 50% of factor activity after 120 min at 37°C. 22 A higher titer signifies a larger amount/more potent inhibitor. For example, a patient with a Bethesda titer of 1 BU/ml would demonstrate 50% factor activity in a 1:1 dilution of patient-to-control plasma, and a BU titer of 10 BU would indicate 50% factor activity in a 1:10 dilution of patient-to-control plasma. Inhibitors are further divided into low or high responding, based on the presence of either a weak or robust response to exogenous factor VIII. Patients who have had a BU assay of 5 BU/ml or more at any time are high responding, and those remaining persistently under 5 BU/ml are low responding. 22
Inhibitor formation is not uncommon. The incidence of factor VIII inhibitors is approximately 30% in patients with severe hemophilia A, and is somewhat less common in patients with moderate or mild hemophilia (3–13% incidence). 23 The overall population prevalence of inhibitors in hemophilia A patients is estimated at 5–7%, and 12–13% in those most severely affected. 23 The majority of patients who do develop inhibitors are high responding; approximately half as many patients develop low-responding inhibitors, while a small number transiently develop, and then subsequently lose their inhibitors. 23
Risk factors for inhibitor formation
Although first described nearly 75 years ago, our understanding of the causes of and risk factors for inhibitor formation remain incomplete. 24 Inhibitor development is a multifactorial event, arising due to a complex interplay between various factors, both patient and treatment related. Perhaps the most well-established risk factor is that of the causative mutation itself. Null mutations, resulting in a total absence of factor VIII, are associated with comparatively higher risk, the commonest of which, intron 22 inversion, is associated with an inhibitor formation rate of approximately 22%. 25 Based on the largest such meta-analysis carried out to date and compared with intron 22 inversion, large mutations bring the largest risk [pooled odds ratio (OR) = 3.6; 95% confidence interval (CI), 2.3–5.7], followed by nonsense mutations (OR = 1.4; 95% CI, 1.1–1.8), intron 1 inversion and splice-site mutations (pooled OR = 0.9; 95% CI, 0.6–1.5 and OR = 1.0; 95% CI, 0.6–1.5, respectively). 25 Patients with small deletions and insertions/missense mutations are at the lowest risk of inhibitor formation (pooled OR = 0.5; 95% CI, 0.4–0.6 and OR = 0.3; 95% CI, 0.2–0.4, respectively). 25 Larger, multidomain nonsense mutations, which result in the total absence of factor VIII, do not allow for primary immune tolerance, while smaller/missense mutations or those that permit the translation of at least some portion of the factor VIII protein, however nonfunctional, may facilitate at least some degree of tolerization. 21
Ethnicity is also an established risk factor, with Black and Hispanic patients being at higher risk, even when genotype is controlled for. 26 There are data to suggest that this difference may arise at least partially due to discrepant haplotypes between recombinant factor VIII products and patients, but further study is needed before such a link can definitively be established. 27
Also theorized to play a role are polymorphic variations in genes fundamental to immunoregulatory control, as mediated by tumor necrosis factor alpha, cytotoxic T-lymphocyte antigen 4, and interleukin 10. 28 Certain human leukocyte antigen class II alleles also appear to be associated with an increased risk of inhibitor formation, a finding with obvious relevance, given the variant haplotype expression patterns expressed among different ethnicities. 28
In addition to patient factors, treatment-related risk factors also exist. Though an area of intense ongoing research and debate, young age (e.g. <6 months) and high intensity of first exposure to factor, implementation of prophylaxis at a latter age, and usage of patient plasma-derived factor VIII concentrates as opposed to recombinant factor VIII products have all been implicated to varying degrees in inhibitor formation.21,29
Management of the hemophilia A patient with inhibitors
The development of an inhibitor is a serious and potentially life-threatening event. When present at high titers, they preclude effective treatment, prophylaxis, or on-demand use of factor VIII. Patients with hemophilia A and inhibitors are not a homogenous group; their treatment is similarly variable. The overall goal of therapy, at least initially, however, is the same: to remove the inhibitor. Immune-tolerance induction (ITI), the practice of regularly and repeatedly infusing factor VIII with the intention of eradicating the immune response over time, is the recommended initial management for patients with severe hemophilia A and high-responding inhibitors.30 –32 Low-responding patients may also benefit from ITI, if the inhibitor appreciably impacts either prophylactic or on-demand factor dosing. 31 Multiple ITI regimens exist, involving a range of factor doses, product sources (e.g. plasma derived or recombinant), timing, and auxiliary treatments. Generally speaking, these regimens are effective in a select cohort of patients (with reported successes of 51–79%); the only consistent predictors of success have been a peak factor VIII inhibitor titer of less than 200 BU, and a titer of less than 10 BU at the time of ITI. 21 Ethnicity, 33 factor dose, 34 factor source, 35 and presence of von Willebrand factor 35 do not appear to impact ITI success, though Hay and DiMichelle 34 did observe a shorter time to immune tolerance in patients receiving higher (200 IU/kg/day) versus lower (50 IU/kg three times/week) doses; via intention-to-treat analysis, immune tolerance was achieved in 39% and 41%, respectively (p = 0.909). 34 The Bonn protocol is a widely used approach; first developed in the 1970s, it utilizes twice-daily coadministration of high-dose factor VIII (e.g. 150 U/kg) and activated prothrombin complex concentrates (aPCC). 36 The Swedish Malmo protocol is a less common regimen that includes intravenous immune globulin and cyclophosphamide given in conjunction with factor VIII. 37 The evidence does not, however, support the use of any specific ITI regimen or adjuvant immune suppression. 38 Immune tolerance induction is ineffective in approximately one third of patients, takes a significant amount of time to achieve efficacious results, and is time, cost, and resource intensive.
In the context of an acute bleeding episode in a patient with an inhibitor, however, a different approach is required, and must take into consideration the severity of the bleed itself, type of response (e.g. low or high responding), and inhibitor titer actually present. Use of factor VIII is the ideal management, and low-responding inhibitor titers may be overcome/neutralized via high-factor doses, achievable both via high-dose boluses or continuous infusion. In those with high-response inhibitors but low titers (e.g. <5 BU/ml) at the time of hemorrhage, this approach may also be effective for short periods of time prior to full anamnestic response. In patients with high-responding titers (e.g. >5 BU/ml), bypassing agents are required; those currently available being activated prothrombin complex concentrate (aPCC) and recombinant-activated factor VII.39,40 Both have been shown effective, and of approximately equal efficacy in the treatment of acute hemorrhage. 39 However, both interpatient and intrapatient response to the two products varies widely, even in the context of similar hemorrhagic episodes.40,41 Prophylactic regimens using either recombinant-activated factor VII 42 or aPCC43,44 have also be used effectively in hemophilia patients with inhibitors. While a comprehensive review and comparison of these products is beyond the scope of this article, several recent high-quality reviews exist. 41 It is in this context that emicizumab, initially known by the designation ‘ACE910,’ was developed.
Emicizumab: bispecific factor IXa-/factor X-directed antibody
Preclinical development
Upon activation by either thrombin (activated factor II, factor IIa) or activated factor X (factor Xa), factor VIII dissociates from von Willebrand factor and acts as a cofactor for factor IXa’s activation of factor X which, in combination with factor Va, activates further thrombin. The structure of factor VIIIa is such that the A1 subunit binds factor X’s heavy chain, factor VIII’s A2 subunit binds the heavy chain of factor IXa, and factor VIII’s light chain (e.g. C1 and C2 subunits) bind factor IXa’s light chain. 45 This binding spatially approximates factor IXa and factor X, dramatically increasing (by several orders of magnitude) factor IXa’s rate of factor X activation 45 [Figure 1(a), 1(b)].
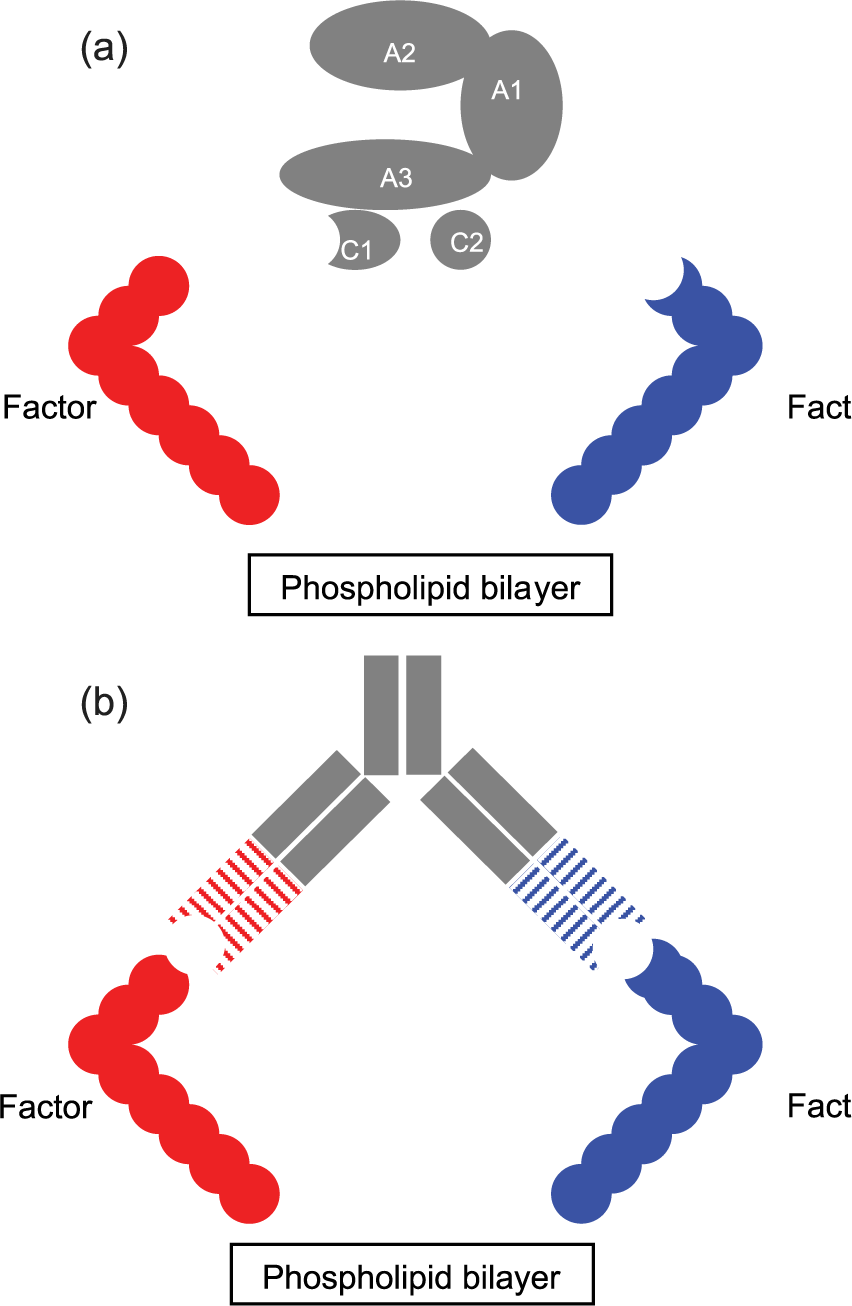
Factor VIIIa’s and emicizumab’s interaction with factor IXa and factor X.
Bispecific antibodies are recombinant, monoclonal antibodies capable of recognizing and binding to two distinct antigenic targets simultaneously. Working from the hypothesis that a bispecific antibody capable of binding factors IXa and X would result in similarly appropriate spatial approximation (and therefore functional equivalency) as factor VIIIa, Kitazawa and colleagues 45 generated hBS23. Briefly, animals were immunized with either human factor IXa or X, facilitating the generation of factor IXa- and factor X-specific monoclonal antibodies. The genes from these animals which encoded the variable regions of the resultant antibodies were inserted into an expression vector, which contained the constant region for human IgG. Human embryonic kidney (HEK) cells (specifically, HEK-293) were cultured following transfection with four expression vectors each, encoding both the light and heavy chains of the factor IXa and factor X antibodies. The ability of each resulting supernatant to generate factor Xa was then evaluated via chromogenic assay. Approximately 40,000 bispecific antibody combinations were screened, resulting in the eventual selection of an antibody with human IgG4 heavy chain and κ light chain, which would subsequently become hBS23 after further humanization. In enzymatic assay testing with purified coagulation factors, hBS23 demonstrated marked factor X activation, but the otherwise-identical, monospecific antibodies to factor IXa or factor X alone did not demonstrate any ability to activate factor X whatsoever; hBS23 did not enhance factor X activation unless both phospholipid and factor IXa were present. Kinetic analysis comparing hBS23 with factor VIIIa showed it to be 1/14th as effective at catalyzing factor X activation; but while hBS23 shortened the activated partial thromboplastin time (aPTT) in factor VIII-deficient plasma to the same extent, irrespective of the presence of inhibitors, recombinant factor VIII’s effect on aPTT was blocked in the presence of inhibitors. Thrombin generation assays demonstrated dose-dependent effects, which persisted in the presence of inhibitors that completely negated recombinant factor VIII’s effects. In subsequent thrombin generation assays, hBS23’s activities at concentrations of 30 nmol/l and 300 nmol/l were equivalent to levels of 0.01 U/ml (1%) and 0.1 U/ml (10%) of recombinant factor VIII, respectively, regardless of whether or not inhibitors were present. 45 Testing in hemophilia A primate models established that a single intravenous bolus administration of 0.3 mg/kg of hBS23 was analogous in hemostatic ability to twice-daily 1 U/kg of recombinant factor VIII, for example, that which was required to maintain a factor activity level of at least 0.01 U/ml (1%). 45 Intravenous pharmacokinetics were consistent with those generally expected of IgG antibodies; hBS23’s half-life was approximately 14 days, with biphasic clearance; subcutaneous bioavailability was 84%, denoting a highly bioavailable route. 45
Sampei and colleagues 46 improved upon hBS23’s pharmacokinetics, solubility, and activity, while reducing its immunogenicity via multidimensional optimization. The resulting product, hBS910 (clinical investigational drug name: ACE910), was twice as effective at catalyzing factor X activation as hBS23 and required only two thirds the concentration to show equivalent activity via thrombin generation assay. Further, its subcutaneous bioavailability was 86%, with a half-life of approximately 21 days and was sufficiently soluble to allow a concentration of 150 mg/ml.
Subsequently, Muto and colleagues47,48 conducted two primate studies using ACE910. The first 47 demonstrated that intravenous ACE910, at a dose of 1–3 mg/kg was equally as efficacious as twice-daily intravenous recombinant factor VIII at a dose of 10 U/kg, in the context of artificially induced subcutaneous/myogenic hemorrhage. The second, 48 acknowledging the significance of hemarthroses in hemophilia A, demonstrated that the usage of weekly subcutaneous ACE910 (an initial bolus of 3.97 mg/kg, followed by 1 mg/kg thereafter) was sufficient to significantly reduce macroscopic joint bleeds and their associated symptomatology, as well as bruising and hematuria.
Kitazawa and colleagues, 45 Sampei and colleagues, 46 and Muto and colleagues47,48 all concluded that clinical efficacy in human patients based on a weekly, subcutaneous dosing schedule could be expected.
ACE910, subsequently designated emicizumab, was specifically designed to mimic factor VIIIa’s activity. Via its ability to interact with and spatially approximate factors IXa and X simultaneously, emicizumab, like factor VIIIa, catalyzes factor X activation. Factor VIIIa and emicizumab are not structurally analogous, however, and important differences exist. Unlike factor VIIIa’s multisite binding, emicizumab interacts with factor IXa and X at only a single site on each protein; it also does so, much less strongly (~100 times and ~6 times weaker interaction, respectively). 49 Emicizumab also binds to both the active and inactive forms of factor IX and X with similar affinity, and does not require activation/proteolysis as factor VIII does; on the other hand, the natural mechanisms by which factor VIIIa is inactivated (e.g. proteolytic degradation by serine proteases) do not apply to emicizumab. 49 The kinetics of IXa/X complex formation are also fundamentally different; in a nonhemophiliac state, the rate of generation of factor VIIIa is itself the limiting step; emicizumab, being in excess, reverses this, and creates a situation in which factor IXa is the limiting reagent. 49 Lastly, perhaps the largest difference is that emicizumab functions exclusively to bring enzyme (factor IXa) and substrate (factor X) into spatial approximation, whereas factor VIIIa, in addition to bridging IXa and X, also aids in factor IXa’s localization to the phospholipid surface, and orientation/stabilization of factor IXa’s active site. 49
Phase I dose-escalation trial
The first human trial of ACE910 was completed in 2015. 50 Using a dose-escalation design, this phase Ia trial randomized a total of 64 healthy, nonhemophilia patients to one of five dosage arms, or placebo. Dosages of 0.001, 0.01, 0.1, 0.3, or 1 mg/kg were given one time via a single subcutaneous injection, and safety profile, immunogenicity, pharmacokinetics and pharmacodynamics evaluated.
ACE910 was well tolerated in all subjects at all doses. No serious adverse events occurred, and other than one case of nasopharyngitis, all adverse events were mild. No clinically relevant changes in factor VIII, IX, or X levels were detected, nor were there any clinically relevant changes in D-dimer, prothrombin time, activated partial thromboplastin time, platelet count, or thrombin-antithrombin III complex, at any dose level. There was, therefore, no suggestion that ACE910 administration might result in a hypercoagulable state.
Peak plasma concentration occurred 1–2 weeks postinjection, with monophasic elimination, and an average half-life of approximately 30 days (range 28.3 ± 4.77 to 34.4 ± 6.55), regardless of dosage group. In factor VIII-depleted plasma, ACE910 demonstrated a dose-dependent increase in thrombin generation and decrease in activated partial thromboplastin time.
Immunogenicity was measured by development of antidrug antibodies (ADAs) which occurred in 4.2% of patients (n = 2), a similar rate, the authors note, to those of other humanized antibodies. Both patients were in the 0.1 mg/kg dose group, and neither’s were immunoglobulin E. One patient’s ADAs were present prior to ACE910 exposure and did not change over the course of the trial; pharmacokinetics/pharmacodynamics were unaffected. The other patient developed ADAs during the trial and had a shortened drug half-life of 9 days, but without alteration of other markers of coagulation function. The authors conclude that hypersensitivity reactions, detrimental effects on simultaneous therapy, and acquired bleeding disorders were all unlikely to develop due to ACE910 exposure, even in the presence of ADAs.
A subsequent phase Ib study by Shima and colleagues, 51 conducted in 18 patients with severe hemophilia A with or without factor VIII inhibitors involved subcutaneous administration of emicizumab on a weekly schedule in a dose-escalation design of 0.3, 1.0, or 3.0 mg/kg. In that study, median annualized bleeding rate fell from 32.5 to 4.4, 18.3 to 0.0, and 15.2 to 0.0, respectively, without evidence of ADA formation or thromboembolic events.
Dosing for the subsequent HAVEN 1 trial (see below) was derived in large part from pharmacokinetic and repeated time-to-event modeling by Yoneyama and colleagues, 52 who characterized the relationship between improved frequency of bleeding and emicizumab pharmacokinetics in the prior studies by Uchida and colleagues 50 and Shima and colleagues, 51 and predicted that a plasma concentration of emicizumab ⩾ 45 µg/ml would result in zero bleeding events for 50% of patients over a period of 1 year. Regimens of 1.5 mg/kg weekly, 3 mg/kg every 2 weeks, or 6 mg/kg every 4 weeks were therefore suggested for evaluation in phase III trials. Interestingly, an overlay of plasma emicizumab concentration versus annual bleeding rate, 52 and factor VIII activity versus annual number of joint bleeds 53 suggests that a plasma emicizumab concentration of ⩾45 µg/ml approximates the bleeding phenotype of patients with mild hemophilia and 12% factor VIII activity level [Figure 2(a), (b)].

(a) Plasma emicizumab concentration versus annual bleeding rate50 and (b) factor VIII activity versus annual number of joint.51
Pivotal clinical trials
HAVEN 1: prophylactic emicizumab versus no prophylaxis in hemophilia A patients with inhibitors
The first such trial of its kind, the HAVEN 1 trial 54 enrolled patients aged 12 years and older with congenital hemophilia A of any severity and high-titer inhibitors (e.g. ⩾5 BU/ml), who were at the time of enrolment being treated with bypassing agents either episodically or prophylactically. Those requiring episodic treatment were randomized 2:1 to prophylactic subcutaneous emicizumab, group A (weekly subcutaneous injection of 3 mg/kg for 4 weeks, followed by a maintenance dose of 1.5 mg/kg weekly thereafter); or control group, group B (no prophylactic emicizumab). Patients needing prophylactic treatment with bypassing agents at the time of enrolment were assigned to group C and received emicizumab prophylaxis in the same fashion as group A. A final cohort, group D, comprised patients unable to enroll in the other groups due to closure, and received emicizumab prophylaxis as described (Figure 3). Episodic use of bypassing agents for breakthrough bleeding was permitted. The primary outcome was the rate of breakthrough bleeds requiring treatment, over at least 24 weeks, between group A and group B; secondary outcomes included characterization of bleeding events, quality-of-life measures, intrapatient comparison of bleeding, safety outcomes, and pharmacokinetics.

HAVEN 1 study design.
109 patients were enrolled, all male, ranging from 12 to 75 years of age (median 28 years), and predominantly of the severe phenotype (n = 102); seven patients suffered from mild or moderate hemophilia A. Median emicizumab exposure was 24 (range 3.0–47.9) weeks. Those receiving emicizumab prophylaxis (group A, n = 35) experienced an annualized bleeding rate of 2.9 events (95% CI 1.7–5.0) compared with 23.3 events (95% CI 12.3–43.9) in those who did not (group B, n = 18); an 87% difference (p < 0.001) (Table 1). In group A, 63% (n = 22) of patients experienced no bleeding events, compared with 6% (n = 1) of those in group B. Patients in group C (n = 24) also experienced a significant improvement in bleeding events, achieving an annualized bleeding rate of 3.3 events (95% CI 1.3–8.1) while on emicizumab, versus a rate of 15.7 events (95% CI 11.1–22.3) while previously receiving prophylaxis with a bypassing agent, a difference of 79% (p < 0.001). In group C, 70.8% (n = 17) of patients experienced no bleeding events while on emicizumab, versus 12.5% (n = 3) using prior therapy (Figures 4 and 5).
HAVEN 1 annualized bleeding rates.

HAVEN 1: prophylactic emicizumab versus no prophylaxis in hemophilia A patients with inhibitors. 52
CI, confidence interval.

HAVEN 1 number of bleeds per patient.

HAVEN 1 intrapatient treated bleeds with emicizumab prophylaxis versus prior bypassing agent prophylaxis.
Adverse events (n = 198, in 103 patients) were predominantly mild to moderate; the commonest were injection-site reactions (n = 28, in 15 patients). Serious adverse events (n = 12, in nine patients) were uncommon; three patients developed thrombotic microangiopathy (TMA) (including one after the cutoff for primary analysis) and two developed thrombotic events (cavernous sinus and superficial thrombophlebitis). All patients developed these complications while receiving infusions of aPCC, and one with TMA also received recombinant factor VIIa. However, a sub-analysis of these events revealed commonality; all received doses of aPCC > 100 U/kg/day, for over 24 h. During the study, 20 patients were treated with aPCC during a total of 78 treatment episodes. Of the episodes in which patients were treated for under 24 h with doses of either ⩽100 U/kg/day (n = 52) or >100 U/kg/day (n = 13), or were treated for over 24 h with doses of ⩽100 U/kg/day (n = 5), no episodes of TMA or thrombosis occurred. In patients who received doses of aPCC > 100 U/kg/day, for more than 24 h (n = 8), five experienced TMA/thrombosis. Two of the five patients subsequently resumed emicizumab, without recurrence. No events occurred in patients treated solely with recombinant factor VIIa, regardless of dosage or timing. No antidrug antibodies were detected in any patient receiving emicizumab, and factor VIII inhibitor titers were apparently unaffected, remaining stable or declining gradually over the course of the study.
The results of the HAVEN 1 and HAVEN 2 trials (see below) led to emicizumab being approved by the US Food and Drug Administration (FDA) in November 2017, for prophylactic use in adult and pediatric patients with hemophilia A and factor VIII inhibitors. 55
An updated analysis of the HAVEN 1 study was presented in December 2017 and included an additional 6 months of follow up. 56 For those patients in arm A (n = 24), median exposure time had increased to 56.29 (range 0.1–74.3) weeks. Median annualized bleeding rate for treated bleeds demonstrated significant improvement from 21.4 (range 15.7–30.8) to 1.2 (range 0.48–2.81); p < 0.0001. A total of 66.7% patients treated with emicizumab prophylaxis experienced no bleeds requiring treatment, versus 8.3% with prior episodic bypassing agents. The annualized bleeding rates were 2.4, 1.0, 3.6 and 2.9 for study arms A, B, C and D, respectively (patients in arm B having been switched to emicizumab prophylaxis after 24 weeks of episodic bypassing agent use), with a median exposure of 40.9 (range 0.1–74.3) weeks and a mean emicizumab trough plasma concentration of 50 µg/ml. These updated results, the authors concluded, showed ongoing evidence of efficacy in the reduction of bleeding rates.
Although patients with planned surgeries were not included in either HAVEN 1 or 2, some unplanned/emergency surgical interventions did occur on a subset of patients. While in HAVEN 1, 17 patients underwent 24 surgical procedures, and 5 patients underwent 5 surgical procedures in HAVEN 2. Prophylactic use of bypassing agents occurred in 31% (n = 9) of procedures. Eight of these patients received a median of single dose of recombinant factor VIIa at a mean dose of 152.81 µg/kg (range 86.54–254.72 µg/kg), and one received aPCC (one dose of 49.78 U/kg). Of the nine surgeries in which bypassing agents were used prophylactically, eight did not experience postoperative bleeding, and one did (a tooth extraction). In the 69% (n = 20) in which they were not used, 30% (n = 6) experienced postoperative bleeds; two were treated with bypassing agents (one tooth extraction, and one arthroscopic synovectomy/debridement/chondroplasty of the right knee). 57
Given the above-noted development of thrombotic microangiopathy/thrombotic events in five patients enrolled in HAVEN 1, the study sponsor issued guidance regarding the usage of bypassing agents in breakthrough bleeding events: it was recommended that all such events be treated with recombinant factor VIIa at an initial dose of ⩽90 µg/kg/day, with avoidance of aPCC if possible. If necessary, it was recommended that aPCC be used at the lowest dose expected to achieve hemostasis, with monitoring for thrombotic microangiopathy/thromboembolic events. 58 Preguideline use of aPCC occurred 63 times in 18 patients, and 15 times in 6 patients postguideline, with reduced doses of aPCC being used (doses of >100 U/kg/day being used in 30.2% and 13.3% of treatment episodes, respectively), and most patients using recombinant factor VIIa when needed (91 treatment episodes in 28 patients preguideline, and 49 treatment episodes in 19 patients postguideline). After issuance of the guideline, one further patient developed thrombotic microangiopathy; that patient had received a dose of >100 U/kg/day for ⩾24 h, as had all preguideline patients who experienced similar such events. The authors therefore concluded that the risk of thrombotic microangiopathy/thromboembolic events could largely be mitigated via adherence to the recommended guidelines for use of aPCC for patients receiving emicizumab. 58
HAVEN 2: prophylactic emicizumab in hemophilia A pediatric patients with inhibitors
The HAVEN 2 trial, 59 which is ongoing, is enrolling those aged less than 12 years (and 12–17 years of age if under 40 kg), with congenital hemophilia A of any severity and high-titer inhibitors (e.g. ⩾5 BU/ml). All enrolled patients are stratified into one of three groups: group A as outlined in the HAVEN 1 study (3 mg/kg for 4 weeks, followed by a weekly maintenance dose of 1.5 mg/kg thereafter); group B (3 mg/kg for 4 weeks, followed by a maintenance dose of 3 mg/kg every 2 weeks); and group C (3 mg/kg for 4 weeks, followed by a maintenance dose of 6 mg/kg every 4 weeks); all results available at this time are in relation to those receiving dosing per group A. The primary outcome measure is the rate of breakthrough bleeding regardless of need for treatment over a period of up to 52 weeks, with secondary outcomes including characterization of bleeding events, quality-of-life measures, intrapatient comparison of bleeding, safety outcomes, and pharmacokinetics.
To date, 88 patients have been enrolled for whom data is available: 95% (n = 57) under 12 years of age, including 3.3% (n = 2) under 2 years of age, and 5% (n = 3) aged ⩾ 12 years and weighing under 40 kg. Of the 57 patients aged < 12 years, 94.7% (n = 54) experienced no bleeds requiring treatment; three patients experienced one bleed each necessitating treatment, and all received recombinant factor VIIa. A total of 65 bleeds occurred in total (including treated bleeds) in 20 patients, and 64.9% (n = 37) experienced no bleeds whatsoever. The median time on study was 9.1 (range 1.6–41.6) weeks; for those patients on study for at least 12 weeks (n = 23), the annualized bleeding rate for treated bleeds was 0.2 events/year (95% CI 0.06–0.62). For the 13 patients for whom such analysis was possible, intrapatient comparison showed a 99% reduction in annualized bleeding rate for treated bleeds following initiation of emicizumab prophylaxis, compared with prior bypassing agents (Table 2).
HAVEN 2 bleeding-related endpoints.

HAVEN 2: prophylactic emicizumab in hemophilia A pediatric patients with inhibitors. 57
CI, confidence interval.
The commonest adverse events were injection-site reaction (16.7%, n = 10) and upper respiratory tract infection (16.7%, n = 10). Seven serious adverse events occurred, none of which were deemed to be secondary to emicizumab (one episode each of appendicitis, oral hemorrhage, catheter infection, indwelling line infection, ocular pain, and two episodes of muscle hemorrhage). No cases of TMA or thromboembolic events were reported. Pharmacokinetics did not vary substantially by weight or age, with trough concentrations of approximately 50 µg/ml maintained.
During HAVEN 2, a single patient developed a clinically significant antidrug antibody to emicizumab, 60 the first and only such instance recorded to date in an overall treatment population of over 600 patients. For that patient, the antidrug antibody resulted in a loss of therapeutic efficacy, with emicizumab being discontinued and prior therapy resumed. In that patient’s case, aPTT, although normal while on emicizumab, became elevated following development of the antidrug antibody; see section ‘Monitoring and Interference with Coagulation Testing.’ Although further evaluation is needed, aPTT may be a reasonable screening test for clearance/neutralization of emicizumab by antidrug antibodies.
HAVEN 3: prophylactic emicizumab versus no prophylaxis in hemophilia A patients without inhibitors
The HAVEN 3 trial [ClinicalTrials.gov identifier: NCT02847637] evaluated patients with hemophilia A of any severity aged 12 years or older receiving either prophylaxis or on-demand factor VIII therapy, who did not have inhibitors. 61 Patients who required on-demand factor VIII were randomized in a 2:2:1 ratio to one of three arms: prophylactic subcutaneous emicizumab at 3 mg/kg for 4 weeks, followed by a weekly maintenance dose of 1.5 mg/kg thereafter (group A); or 3 mg/kg for 4 weeks, followed by a maintenance dose of 3 mg/kg every 2 weeks (group B); or no emicizumab prophylaxis (e.g. continuation of prior on-demand factor VIII dosing), group C. Patients requiring prophylactic factor VIII dosing were instead switched to a regimen of prophylactic subcutaneous emicizumab at 3 mg/kg for 4 weeks, followed by a weekly maintenance dose of 1.5 mg/kg thereafter (group D). Factor VIII treatment was permitted for any breakthrough bleeding, in all cohorts. Primary outcome measure was the number of bleeds over time, with secondary outcomes including characterization of bleeding events, quality-of-life measures, intrapatient comparison of bleeding, safety outcomes, and pharmacokinetics.
Results for the HAVEN 3 trial were presented in May 2018. 62 A total of 152 patients were enrolled. The annualized bleeding rates for patients in groups A, B, and C (95% CI) were 1.5 (0.9–2.5), 1.3 (0.8–2.3), and 38.2 (22.9–63.8), respectively; no bleeds were experienced in 55.6%, 60.0%, and 0.0%, respectively. Compared with group C, this represents a 96% and 97% reduction in bleeding rate for group A and group B, respectively. Comparison between group A and C, and group B and C were both statistically significant, favoring emicizumab (p < 0.0001). Additionally, patients in group D, who switched from prophylactic factor VIII to emicizumab, achieved a 68% reduction in intraindividual annualized bleeding rate (n = 48; p < 0.0001), from 4.8 (3.2–7.1) to 1.5 (1.0–2.3) events per year. By far the most common adverse events were injection-site reactions (occurring in 38 patients); arthralgia, nasopharyngitis, upper respiratory tract infection, and headache were also all experienced by at least 5% of patients; no thrombotic microangiopathy, thrombotic events, or deaths occurred. Overall, 14 serious adverse events occurred, one of which was deemed to be related to emicizumab therapy. In patients coexposed to both emicizumab and factor VIII (n = 64), no serious adverse events occurred, nor were any de novo factor VIII inhibitors or antidrug antibodies detected.
HAVEN 4: emicizumab given every 4 weeks in patients with hemophilia A
The HAVEN 4 trial 63 is a single-arm phase III study evaluating the use of emicizumab administered prophylactically to patients aged ⩾ 12 years with congenital hemophilia A with or without inhibitors. This study comprised a pharmacokinetic phase, the goal of which is to assess pharmacokinetics, efficacy, and safety of subcutaneous emicizumab administered at a dose of 6 mg/kg every 4 weeks for at least 24 weeks, followed by an expansion phase, which will additionally include a loading dose of 3 mg/kg weekly for 4 weeks followed by 6 mg/kg every 4 weeks.
A total of 48 patients were included in HAVEN 4; 63 7 in the pharmacokinetic phase, and 41 in the expansion phase (patients treated in the pharmacokinetic phase also moved on to the expansion phase). Mean annualized bleeding rate for all bleeds was 2.1 (95% CI 0.0–5.9); 29.3% of patients experienced no bleeds (95% CI 16.1–45.5). Mean annualized bleeding rate for treated bleeds was 2.5 (95% CI 1.4–4.3), with a median annualized bleeding rate for treated bleeds of 0.0 (interquartile range 0.0–2.1); 56.1% (95% CI 39.7–71.5%) of patients experienced no bleeds requiring treatment, and 90.2% (95% CI 76.9–97.3%) experienced 3 or fewer treated bleeds. A single serious adverse event was reported (grade 3 hypertension in a single patient with a history of the same). The most common adverse events were injection-site reactions, experienced by 22% of patients. No thrombotic microangiopathy, thromboembolic events, or deaths occur.
Ongoing trials
Several additional studies are either open or expected to open in the near future:
The study of emicizumab prophylaxis in participants with hemophilia A and inhibitors undergoing minor surgical procedures [ClinicalTrials.gov identifier: NCT03361137] is a phase IV, single-arm, open-label, multicenter study that intends to evaluate the safety of minor surgical procedures in patients with hemophilia A and inhibitors, who are receiving emicizumab prophylaxis. The researchers will assess this by evaluating the proportion of patients with varying degrees of surgical-site bleeding, and their resultant need (or lack thereof) of bypassing agents. This study opened in January 2018, and completion is anticipated by June 2019.
The efficacy, safety, and pharmacokinetic study of prophylactic emicizumab versus no prophylaxis in hemophilia A participants (HAVEN 5) [ClinicalTrials.gov identifier: NCT03315455] is a phase III, randomized, open-label, multicenter study intended to assess emicizumab in patients with hemophilia A, regardless of inhibitor status. Patients receiving either regular factor VIII or regular bypassing agents will be randomized to one of three arms, in a 2:2:1 ratio: (a) 3 mg/kg emicizumab, subcutaneously once per week for 4 weeks, followed by 1.5 mg/kg emicizumab once per week; (b) 3 mg/kg emicizumab, subcutaneously once per week for 4 weeks, followed by 6 mg/kg emicizumab every 4 weeks; or (c) no emicizumab prophylaxis. It is open to patients 12 years of age and older with hemophilia A, both with and without inhibitors. The study goal is to assess the number of bleeds experienced, as well as how these bleeds vary from the patient’s baseline. This study is anticipated opening in April 2018 and being completed by February 2020.
A study to evaluate the safety and tolerability of prophylactic emicizumab in hemophilia A patients with inhibitors (STASEY) [Clinical-Trials.gov identifier: NCT03191799] is a phase IIIb, single-arm, open-label, multicenter study intended to evaluate emicizumab’s safety profile in patients with hemophilia A and inhibitors (of any titer) against factor VIII. Patients 12 years of age and older are eligible and will be administered an initial prophylactic subcutaneous emicizumab dose weekly for 4 weeks and then weekly maintenance doses over a 2-year period. The primary aim is to assess incidence and severity of adverse events, with secondary goals of assessing pharmacokinetics, anti-emicizumab antibody development, quality of life, and bleeding frequency. This study opened in September 2017 and completion is anticipated in September 2020.
A study to investigate the pharmacokinetics, safety, and tolerability of emicizumab in healthy Chinese volunteers [ClinicalTrials.gov identifier: NCT03380780] is a phase I, open-label, single-center study being performed at Peking University Third Hospital in Beijing, China. The researchers aim to assess the safety, tolerability, and pharmacokinetics of emicizumab in healthy Chinese patients, following a single 1 mg/kg subcutaneous dose. This study opened in January 2018 and completion is anticipated by July 2018.
In addition, one expanded-access (compassionate use) program exists: ‘An expanded-access program of emicizumab in participants with hemophilia A with inhibitors’ [ClinicalTrials.gov identifier: NCT03154437] is an open-label, multicenter expanded access program, intended to allow eligible patients access to emicizumab prior to commercial availability. Patients 12 years of age and older, with hemophilia A and high-titer (e.g. ⩾5 BU) inhibitor are eligible for inclusion.
Emicizumab: important considerations and adverse events
Thrombotic microangiopathy, thromboembolism, and adverse events
Emicizumab carries a boxed label warning regarding TMA and thromboembolism due to the occurrence of these events in a subset of patients.69 All patients who developed these complications received aPCC at a dose of over 100 U/kg/24 h, for a duration of more than 24 h. Patients who developed TMA presented with thrombocytopenia, microangiopathic hemolytic anemia, and acute kidney injury. In all cases, discontinuation of aPCC resulted in improvement within 1 week, and full resolution of the TMA. No patients who developed thromboembolic events required anticoagulation, and improvement or resolution was documented in all cases within 1 month of aPCC being stopped. While aPCC may still be given to patients receiving emicizumab, the benefits and risks of doing so must be carefully considered. If features of either TMA or thromboembolism develop, emicizumab and aPCC should both be stopped, and the condition managed as indicated. Emicizumab should be resumed following resolution, on a case-by-case basis.
The most common adverse event experienced during clinical trials was injection-site reaction, experienced by 19% of patients, and including pain, hematoma, bruising, erythema, induration, pruritus, or rash. Erythema (7.4%), pruritus (5.3%) and pain (5.3%) were the most common, and all site reactions were of mild to moderate severity only, with 88% resolving without intervention. Other adverse events included headache (15%), arthralgia (10%), pyrexia (7%), diarrhea (6%), and myalgia (5%).
Monitoring and interference with coagulation testing
Emicizumab enhances the generation of factor Xa, as discussed above, and does so by behaving like constantly activated factor VIIIa. Coagulation assays based on the intrinsic pathway (such as the aPTT) which measure the time needed for activation of factor VIII by thrombin will therefore yield artificially shortened times, as emicizumab is not activated by thrombin, but rather, is always ‘on.’ All single-factor assays based upon aPTT will thus be affected, as will activated clotting time, clotting-based Bethesda assays for factor VIII inhibitor titers (resulting in a false negative), and aPTT-based protein C resistance. As such, these tests should not be used, including in determining adequacy of dosing, anticoagulation, or monitoring of activity. 64 As the half-life of emicizumab is approximately 28 days, lab results may be impacted for up to 5–6 months following the last dose. On the other hand, chromogenic and immune-based single-factor assays (with the exception of factor VIII chromogenic assays) are unaffected by emicizumab; tests which do not measure the intrinsic coagulation pathway are likewise unaffected, including thrombin time and one-stage prothrombin-time-based factor assays. Regarding chromogenic factor VIII assays, these may be manufactured using bovine or human proteins. Those with bovine coagulation factors are not sensitive to emicizumab, and therefore may be reliably used to assay native or exogenous factor VIII activity, or factor VIII inhibitors. In contrast, those chromogenic assays manufactured using human coagulation factors are sensitive to emicizumab and may overestimate factor VIII activity. Additionally, a commercially available assay has recently been developed to allow measurement of emicizumab activity. 65 This technique utilizes a modified one-stage aPTT-based factor VIII assay, with increased predilution of the sample performed, calibrated against emicizumab. The clinical utility and benefit of this assay is not fully known at this time.
Conclusions and remaining questions
Despite the large number of ongoing trials, some outstanding questions do remain, however, including the role of emicizumab in previously untreated patients, in particular, in the prevention and management of serious bleeding events, the impact of emicizumab on the development of inhibitors/inhibitor kinetics while on emicizumab, and the long-term effects on bone/joint health in hemophilia patients. Additionally, an effective assay to monitor emicizumab levels and treatment efficacy has not yet been developed. These questions will likely be answered as emicizumab comes into wider use within the hemophilia A community. However, it seems certain that emicizumab is poised to dramatically alter the treatment landscape for patients with hemophilia A and inhibitors. Trials in patients without inhibitors are ongoing, but preliminary assessments strongly suggest that it will be a game-changing drug for these patients as well, and for the physicians who care for them.