
Abstract
Background:
Nilotinib, a second-generation tyrosine kinase inhibitor (TKI), is approved for the treatment of patients with chronic myeloid leukemia (CML) in many countries, including Taiwan. Though a number of controlled clinical trials have demonstrated the safety and efficacy of nilotinib, studies assessing the safety and efficacy of nilotinib in routine clinical practice are limited.
Methods:
The current study was an open-label, single-arm study conducted across 12 centers in Taiwan in adult patients with CML in chronic or accelerated phase with confirmed Ph+ chromosome (or BCR-ABL) and resistant or intolerant to one or more previous TKIs. The primary objective was to collect the long-term safety data in patients treated with nilotinib 400 mg, twice daily for up to 2 years.
Results:
The study enrolled 85 patients with CML, including 76 in the chronic phase (CML-CP) and 9 in the accelerated phase (CML-AP). Overall, 1166 adverse events (AEs) were reported in 80 patients (94.1%), of which 70 AEs (6%) in 28 patients (32.9%) were serious and 336 AEs (28.8%) reported in 60 patients (70.6%) were drug-related. Common drug-related AEs were thrombocytopenia (21.2%), increased alanine aminotransferase (21.2%) and pruritus (17.7%). Of the 85 patients, 19 switched from imatinib due to intolerance – AEs were resolved in 16 of these 19 patients (84.2%). By 24 months, the cumulative rates of complete cytogenetic response (CCyR), major molecular response (MMR), MR4.0 (BCR-ABL1IS ⩽0.01%) and MR4.5 (BCR-ABL1IS ⩽0.0032%) were 75.3, 56.8, 16.2 and 7.4%, respectively. Patients with CML-CP at baseline had higher overall survival (OS) and progression-free survival (PFS) than those with CML-AP.
Conclusion:
This is the first study that demonstrated that nilotinib is effective and well-tolerated in patients resistant or intolerant to imatinib in the real-world setting in Taiwan, reflecting effective management of CML by physicians under routine clinical practice in Taiwan.
Introduction
Chronic myeloid leukemia (CML) is a clonal disease of the hematopoietic stem cells associated with a specific chromosomal translocation resulting in the Philadelphia (Ph) chromosome. The fusion BCR-ABL1 gene thus formed leads to constitutive expression of the BCR-ABL1 kinase resulting in the manifestation of CML.1,2 The development of imatinib mesylate (Glivec®, Novartis AG), a selective tyrosine kinase inhibitor (TKI) of BCR-ABL, has revolutionized the treatment for Ph+ CML, and imatinib is now considered the standard of care for patients with Ph+ CML, especially in the chronic phase. 3 The long-term safety and efficacy of imatinib were demonstrated in the pivotal International Randomized Study of Interferon and STI571 (IRIS) trial [ClinicalTrials.gov identifier: NCT00006343], with an estimated overall survival (OS) rate of over 80% after 8 years of treatment and 83.3% after 10 years of treatment.4–6 However, it was observed that approximately 20–30% of patients developed resistance or intolerance to imatinib. 7
Nilotinib (Tasigna®, Novartis AG), a second-generation oral TKI, was initially developed to treat patients with CML who are resistant or intolerant to imatinib.
8
Nilotinib is effective and well-tolerated in these patients. In patients with chronic phase CML (CML-CP) receiving nilotinib as second-line therapy, major cytogenetic response (MCyR) was achieved in 59% of patients and complete cytogenetic response (CCyR) in 44% of patients, and the estimated OS was approximately 80% at 48 months.9,10 Based on the results from the pivotal registration trial CAMN107A2101 [ClinicalTrials.gov identifier: NCT00109707], nilotinib was approved in 2007 for the treatment of imatinib-resistant CML-CP or for patients with CML in the accelerated phase (CML-AP). Subsequently, in 2010, it was approved by the US Food and Drug Administration (FDA), European Medicines Agency (EMA), and health authorities of several countries globally for the treatment of newly diagnosed patients with CML based on the results from pivotal phase III ENESTnd study [ClinicalTrials.gov identifier: NCT00471497].
11
In 2008, Taiwan gained regulatory approval for nilotinib as a second-line treatment for patients with CML. However, the pivotal registration trial had enrolled only one patient from Taiwan.
9
Following the approval of nilotinib in Taiwan, there was clear interest from local physicians to gather more safety data in a larger population of Taiwanese patients with CML. To further examine the safety and efficacy of nilotinib as second-line treatment, a noninterventional, multicenter, observational study,
Methods
Study design, treatment and patients
NOVEL was a noninterventional observational study conducted in 12 centers across Taiwan. The primary objective of the study was to collect long-term safety data on nilotinib treatment in patients with CML-CP or CML-AP during routine clinical practice in Taiwan. In addition, the study also evaluated efficacy parameters including hematological, cytogenetic, and molecular responses (MR). During the study period of 2 years, efficacy and safety data were collected from patients who were treated with nilotinib 400 mg twice daily under routine clinical practice.
Adult patients with CML-CP or CML-AP who were resistant or intolerant to at least one first-line TKI therapy for CML and with an Eastern Cooperative Oncology Group (ECOG) performance status ⩽2 were included in the study. Patients previously treated with dasatinib, who met the definition of imatinib resistance or intolerance, were also enrolled. Patients in the blast phase of CML or with a prior history of blast phase or with previously documented T315I mutation were excluded from the study. In addition, patients with a significant risk of developing prolonged QTc or with a clinically significant uncontrolled heart disease, with hereditary metabolic conditions, with other concurrent malignancies or who were pregnant or breastfeeding were also excluded. All patients were required to provide a written informed consent. The study protocol and amendments were reviewed by an ethics committee and institutional review board at each participating center, and the study complied with the latest Declaration of Helsinki, Good Clinical Practices and Good Postmarketing Study Practices.
Assessments
The safety and clinical response data were collected and recorded under routine clinical follow up. Safety assessments, including hematological and biochemical laboratory investigations, were performed at baseline, every month for the first 3 months, and quarterly thereafter. Adverse events (AEs) were recorded in terms of their frequency as well as the number of patients with AEs according to the Common Terminology Criteria for Adverse Events (CTCAE), and were monitored continuously. The relationship of AEs to the drug was assessed by the judgment of the investigator and was based on standard criteria in assessing the relationship of AEs in clinical studies. In addition, the safety and clinical response data were also collected retrospectively for up to 1 year for patients who had already commenced nilotinib treatment at the time of study entry.
At baseline, the reason for switching to nilotinib was collected. It was not mandatory to collect the cytogenetic and MR data at baseline due to the observational nature of the study, and the patients may have had cytogenetic response and MR at baseline. The hematological, cytogenetic and MR parameters were collected every 3 months according to the European Leukemia Net (ELN) guidelines for the management of patients with CML. 12 Complete blood and differential cell counts were used to assess the hematologic response, and were conducted at local laboratories. Complete hematologic response (CHR) was defined as platelet count <450 × 109/L, white blood cell (WBC) count <10 × 109/L, without immature granulocytes, and with <5% of basophils, and nonpalpable spleen.13,14 Cytogenetic response was evaluated using standard bone marrow assessments based on the percentage of Ph+ metaphases found in 20 or more metaphase cells in a bone marrow sample. 13 MR assessments were conducted at the central laboratories in Taiwan using quantitative real-time reverse transcriptase polymerase chain reaction (RQ-PCR) to quantify the BCR-ABL1 transcripts using peripheral blood samples. 15 Major molecular response [MMR; BCR-ABL1 on the international scale (IS) ⩽0.1%], MR 4.0-log reduction from a standardized baseline (MR4.0; BCR-ABL1IS ⩽0.01%) and MR 4.5-log reduction from a standardized baseline (MR4.5; BCR-ABL1IS ⩽0.0032%) were also assessed. During the study, certain exploratory outcomes such as the patients’ baseline BCR-ABL mutations and/or secondary mutations were also recorded when performed by the investigators.
Statistical analysis
The study did not have a formal sample size calculation, as this was a noninterventional, observational study. Continuous variables were summarized by mean, median, standard deviation (SD), minimum, maximum and 95% confidence intervals (CIs), and the categorical variables were summarized by counts and percentages in the frequency table. The time-related variables were analyzed using the Kaplan–Meier (KM) method. To estimate the response rates, the KM method was applied to impute missing data. When treatment response was not available at a certain time point, response reported at the previous visit was adopted for estimation. In addition, retrospective data collection of the past year was allowed in this study. The time points for the return visits were adjusted to reflect the actual treatment duration in each patient. The duration of response was calculated in patients who achieved a complete or partial hematological remission and was measured from the date of first documented response to the date of loss of response or death due to any cause, whichever occurred first. If a patient experienced no response during the study period, time to response was censored at the time of the last follow up. All statistical assessments were evaluated for a two-sided interval under a significance level of 0.05.
Results
Patients
A total of 85 patients were enrolled from 27 July 2010 to 30 December 2011 across 12 centers in Taiwan and were followed up for 2 years. At the data cutoff on 13 January 2014, 54 of 85 patients (63.5%) enrolled had completed the study. Overall, 31 patients (36.5%) had discontinued treatment and study, predominately due to unsatisfactory therapeutic effect (n = 14; 16.5%), followed by withdrawal of consent (n = 5; 5.9%), development of intolerable toxicities (n = 4; 4.7%), death (n = 3; 3.5%), development of nilotinib resistant F359C and E459K mutations (n = 1; 1.2%), poor compliance (n = 1; 1.2%), pregnancy (n = 1; 1.2%), administrative problems (n = 1; 1.2%) and switching to another drug (n = 1; 1.2%). The median age of the study population was 47 years (range, 21–85 years), of whom 48 were male (56.5%). About 89% of the patients enrolled were in CML-CP (Table 1). At enrollment, the median time from initial CML diagnosis was 20.3 months (range, 1.4–287.7 months). Of the 85 patients, 4 did not report any history of comorbid conditions. The comorbid conditions of interest include hematological conditions (including anemia not related to iron deficiency or chronic disease, thrombocytopenia, leukopenia, lymphopenia, decrease in platelet count and decrease in WBCs) in 18 patients, hypertension in 20 patients, diabetes in 12 patients, hyperlipidemia in 3 patients, cardiovascular events in 9 patients, chronic obstructive pulmonary disease in 3 patients and pleural effusion in 3 patients. Mutation analysis was performed for 33 patients at baseline, which revealed 7 mutations (E450G, E543A, F317L, F486S, G250E, M244V and M351T; n = 1 each); CHR was achieved in 61 patients (71.8%).
Patient characteristics.

CHR, complete hematologic response; CML, chronic myeloid leukemia; TKI, tyrosine kinase inhibitor; WBC, white blood cells.
Poor compliance.
One patient had F317L mutation and another patient had anterior wall ischemia.
Before entering the study, all patients (100%) had received imatinib, with a mean ± SD dose of 473 ± 153 mg/day at the last treatment; 19 patients (22.4%) had also received dasatinib, with a mean ± SD dose of 98.9 ± 21.9 mg/day at the last treatment. Of the 19 patients treated with both imatinib and dasatinib, 17 received imatinib first followed by dasatinib, and 2 received dasatinib first followed by imatinib. Approximately, 70% of patients switched from imatinib due to lack of response and the rest switched due to intolerance or due to both; among the patients switching from dasatinib, the proportion of patients switching due to lack of response and due to intolerance was similar (Table 1).
Adverse events
The safety population comprised all enrolled patients who had received at least one dose of nilotinib. During the 2-year observational period, 80 patients (94.1%) experienced at least one AE, while 60 patients (70.6%) experienced drug-related AEs. A total of 1166 AEs were reported, of which 336 events (28.8%) were drug-related. The most commonly reported nonhematological AEs (regardless of relationship to study drug) were cough (34.1%), increase in alanine aminotransferase (ALT; 24.7%), constipation (23.5%), pruritus (22.4%), dizziness (22.4%), upper respiratory tract infection (21.2%), pyrexia (18.8%), rash (17.7%), headache (15.3%), pain in extremity (15.3%), rhinorrhea (15.3%), increase of bilirubin (14.1%), insomnia (12.9%), palpitation (12.9%), chest discomfort (12.9%), back pain (12.9%), diarrhea (11.8%), fatigue (10.6%), chest pain (10.6%) and increase of aspartate aminotransferase (AST; 10.6%) (Table 2). The frequent hematological AEs included thrombocytopenia in 20 (23.5%), anemia in 18 (21.2%), neutropenia in 7 (8.2%) and leukopenia in 5 patients (5.9%), respectively. Grade 3 or 4 AEs were uncommon, and mostly comprised hematological laboratory abnormalities, including thrombocytopenia in eight patients (9.4%), anemia in five patients (5.9%) and neutropenia in three patients (3.5%). Some of the more frequently reported grade 3 or 4 nonhematological AEs included lipase elevation and pneumonia in four patients (4.7%) each and acute respiratory failure in three patients (3.5%), while pyrexia, AST elevation and pleural effusion were noted in two patients (2.3%) each. Cardiac disorders were reported in 16 patients (18.8%), including palpitations in 11 patients (12.9%), and angina pectoris, myocardial ischemia and sinus bradycardia in two patients (2.4%) each. Myocardial infarction, arrhythmia, cardiac failure, congestive cardiac failure, cardiomegaly, dizziness and mitral valve incompetence were reported in one patient (1.2%) each.
Nonhematological AEs (>10% incidence).
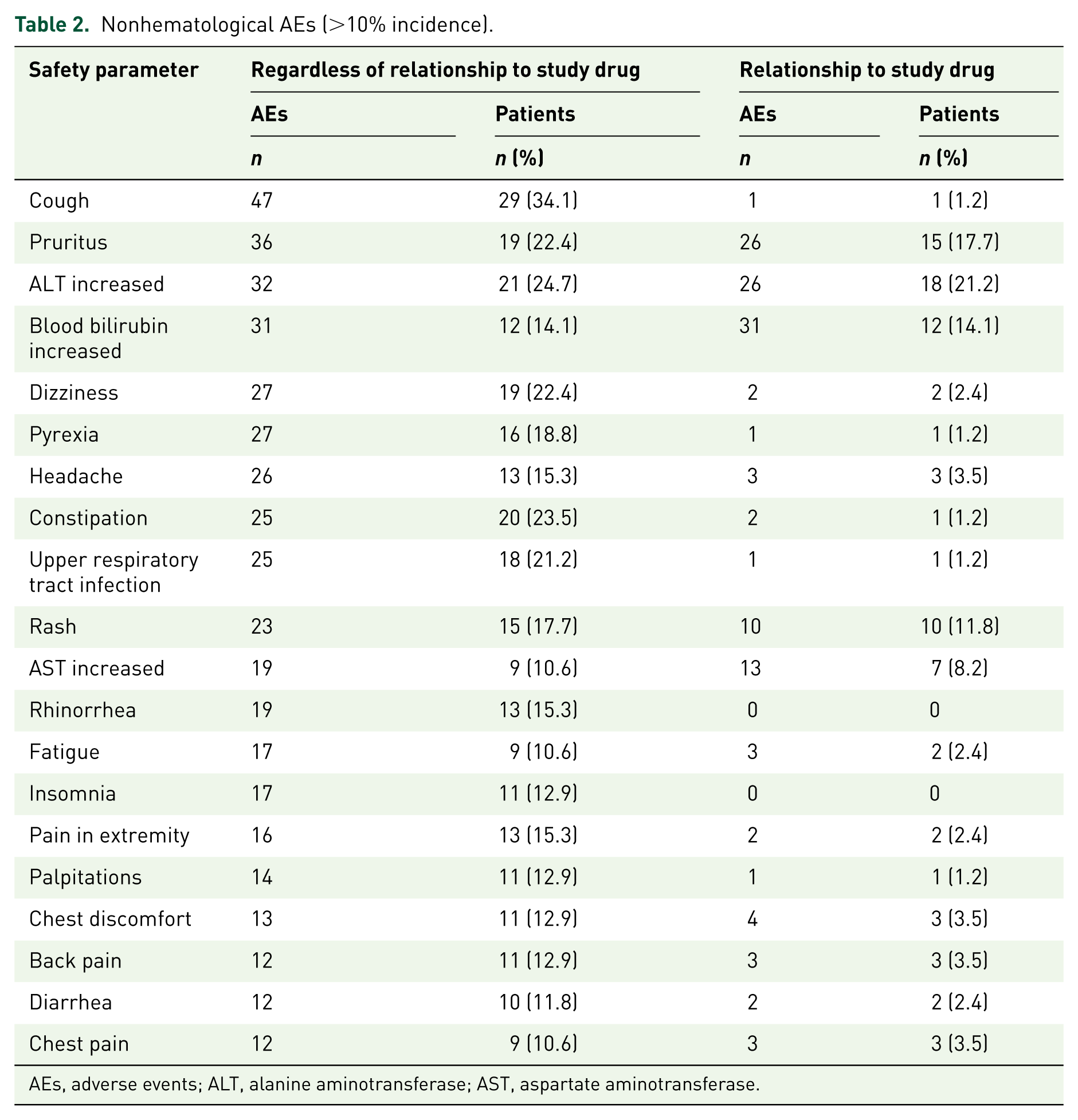
AEs, adverse events; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Of the 1166 AEs reported, 336 were considered to be related to nilotinib, including 85 hematological and 251 nonhematological AEs reported in 60 patients (70.6%). Common drug-related AEs were increase in ALT and thrombocytopenia in 21.2% of patients each, respectively, followed by pruritus (17.7%), increase in blood bilirubin (14.1%), anemia (14.1%) and rash (11.8%) (Table 3). Overall, 25 hematological and 16 nonhematological AEs, reported in 10 (11.8%) and 8 patients (9.4%), respectively, were graded 3 or 4 in severity. Dose reduction or interruptions were reported in 26 patients.
Drug-related hematologic laboratory abnormalities and nonhematologic AEs by preferred term and biochemical abnormalities (incidence rate >5% of ITT population).

AEs, adverse events; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ITT, intent to treat.
Nilotinib was well-tolerated in patients who had switched to nilotinib due to intolerance to imatinib (Table 4). In total, 25 patients (29.4%) had switched to nilotinib due to AEs, of whom 6 had switched due to unknown AEs. Of the 19 patients who switched to nilotinib due to known AEs with imatinib, AEs were resolved in 16 patients (84.2%). One instance each of cytopenia, allergy and arthralgia experienced with imatinib were not resolved after switching to nilotinib.
Cross-tolerance AE rates between imatinib and nilotinib.

AEs, adverse events.
Serious adverse events and deaths
Of the 1166 AEs, 70 events (6.0%) were serious AEs (SAEs), reported in 28 patients (32.9%). Eight drug-related SAEs were reported, which included jaundice, grade 3 QTc interval prolongation, poorly controlled diabetes mellitus, sinus bradycardia, chronic hepatitis, acute recurrent left thalamus infarction, infarction at left basal ganglia and corona radiata, and palpitations. In addition, five deaths were reported during the study. One death caused by cardiopulmonary failure was suspected to be related to nilotinib. Four other deaths, respectively caused by acute myelogenous leukemia, traffic accident, chronic obstructive pulmonary disease with exacerbation and diffused subarachnoid hemorrhage, were not suspected to be related to nilotinib. Two additional deaths caused by pneumonia and sepsis were reported during follow up, but were not suspected to be related to nilotinib.
Efficacy
The cumulative CHR, CCyR and MMR rates by 3 months were 72.9%, 28.2% and 11.9%, and none of the patients had achieved MR4.0 or MR4.5 in the overall population. By 24 months, 94.5%, 75.3%, 56.8%, 16.2% and 7.4% achieved CHR, CCyR, MMR, MR4.0 and MR4.5, respectively (Figure 1(a)). The median time to achieve CHR, CCyR and MMR were 2.5 months (95% CI 1.7–3.0), 8.9 months (95% CI 5.7–12.0) and 20.8 months (95% CI 11.9–32.4), respectively (Figure 1(b)). In the 76 patients with CML-CP at baseline, by 24 months the cumulative rates of CHR, CCyR and MMR were 93.9%, 77.0% and 56.5%, respectively, which were similar to those of the overall population. Of the nine patients who were in CML-AP at study entry, the cumulative rates of CHR, CCyR and MMR were 100%, 58.3% and 58.3% respectively by 24 months. Of the 22 patients (25.9%) who were not in CHR at baseline, 21 (95.5%) achieved CHR and 15 (68.2%) achieved CCyR and MMR by 24 months. The median time to achieve CHR and CCyR was 3.1 months (95% CI 1.0–10.0) and 11.5 months (95% CI 5.0–21.6), respectively, which was longer than that in the overall population. However, the median time to achieve MMR was shorter in these patients compared to the overall population (15.4 months; 95% CI 9.6–24.3). Patients with confirmed BCR-ABL1 mutations also showed delayed time to response. The median time to achieve CHR and MMR was 9.0 and 37.0 months in patients with mutations, respectively, compared to 2.3 and 16.9 months, respectively, in patients without mutations.

Response rates in all patients over time (ITT). (a) Cumulative response rates in all patients by time. (b) Time to response.
Better response outcomes were seen in patients who were intolerant to imatinib compared to those who were resistant to one previous TKI; however, some of the patients who switched to nilotinib due to intolerance to imatinib may have already achieved CHR, CCyR or MMR at baseline. Furthermore, patients who were resistant to both imatinib and dasatinib demonstrated delayed median time to response compared to patients resistant only to imatinib (Supplementary Figure 1). Among the 50 patients resistant to imatinib only, all achieved CHR after a median time of 2.4 months, whereas only three of four patients resistant to two TKIs achieved CHR after a median time of 13.5 months. The rates of CCyR and MMR by 24 months were lower in patients resistant to two TKIs, who demonstrated longer median time to achieve these responses compared to patients resistant to imatinib only (Supplementary Figure 1). Of the 24 patients who switched from imatinib due to intolerance, all achieved CHR by 24 months with a median time to response of 2.2 months, 20 patients (83.3%) achieved CCyR with a median time to response of 3.7 months, and 17 (70.8%) achieved MMR with a median time to response of 12 months.
Early molecular response (EMR) is of value to predict the long-term outcome of target treatment. 16 As this was an observational study, limited numbers of patients were eligible for determination of EMR. In the study, 27 patients were eligible for EMR analysis at 3 months. Of these, 20 patients achieved EMR (BCR-ABL1IS ⩽10%); 19 of these 20 patients achieved CCyR (mean time, 7.0 months), while 13 of 20 patients achieved MMR with a mean time of 11.8 months, 4 achieved MR4.0 with a mean time of 14.3 months, and 1 patient achieved MR4.5 at 18.8 months. Of the 7 patients who did not achieve EMR at 3 months, all achieved CHR with a mean time of 2.0 months, and 1 patient achieved CCyR at 9.1 months, and MMR at 11.9 months; by 24 months, no patient had achieved MR4.0 or MR4.5. Among the 35 patients eligible for 6-month EMR analysis, 25 achieved an EMR. Among these, 23 patients achieved CCyR (mean time, 6.9 months), 18 patients achieved MMR (mean time, 11.9 months), 6 patients achieved MR4.0 (mean time, 14.3 months) and 2 patients achieved MR4.5 (mean time, 18.4 months). Overall, 10 patients did not achieve EMR at 6 months, 9 achieved CHR with a mean time of 1.8 months, 3 achieved MMR with a mean time of 14.1 months, and no patients achieved MR4.5 if without EMR at 6 months.
Of the patients with mutations at baseline, three patients with mutations – G250E, F486S and E543A – had CHR at baseline. By the end of the study the patient with mutation G250E had achieved MMR, and the patients with F486S and E543A had maintained CHR. Four patients with mutations E450G, M244V, M351T, F317L did not have CHR at baseline. Among these, the patient with E450G achieved MMR, while the patient with M244V achieved CHR. The patient with mutation M351V, though unable to achieve CHR by the end of the study, completed the study, while the patient with F317L discontinued due to being unable to achieve CHR. Additionally, among the patients tested for mutations during the study, four patients developed mutations: two had mutation T3151, one had F359 and E459K, and the other had F317L. All four patients discontinued the study due to lack of response.
Disease progression and overall survival
The median OS or progression-free survival (PFS) of the study cohort was not reached during the 2-year observational period. Disease progression was seen in eight patients (9.4%), included four patients each with CML-CP or CML-AP at baseline (Figure 2). At the end of the 2-year observational period, seven patients (8.2%) had died after a mean period of 41.7 months. The median time to death was 42.3 months for six patients who had achieved CHR as their best response before death. One patient had reached MR4.5 but died at 21.8 months due to chronic obstructive pulmonary disease with exacerbation, not suspected to be related to the study drug.

Overall survival and progression-free survival (ITT population). (a) Overall survival in all patients. (b) Overall survival in patients by disease state. (c) Progression-free survival in all patients. (d) Progression-free survival in patients by disease state.
As shown in Figure 2, OS and PFS were significantly higher in patients with CML-CP at baseline compared to those with CML-AP at baseline. The median OS and PFS were not reached for patients with CML-CP at baseline (Figure 2). In patients with CML-AP at baseline, the median OS was 42.3 (95% CI 4.4–42.3) months, and the median PFS was 42.3 (95% CI: 3.3–42.3) months. A Cox regression model was applied to identify predictors for OS and PFS, such as reasons for switching to nilotinib, gender, age, CHR status at baseline and the clinical phase of CML at screening. Among all the predictors tested, only the phase of CML was identified as a significant predictor for both OS and PFS [hazard ratio (HR) for OS, 18.49; 95% CI 3.38–101.19; p = 0.0008; HR for PFS, 9.6826; 95% CI 2.41–38.90; Supplementary Table 1].
Discussion
The NOVEL study evaluated the safety and efficacy of nilotinib as a second-line or third-line treatment for patients with CML in Taiwan. Since this study collected data from the real-world setting, it provides valuable information regarding the management of these patients by clinicians in Taiwan. Although Taiwan had participated in the pivotal registration study and the Expanding Nilotinib Access in Clinical Trials (ENACT) study, the patient population from Taiwan evaluated in these studies was <50,9,17–19 and the studies did not specifically assess the outcomes in these patients. Thus, the present study provides insights into whether differences in ethnicity influence treatment and outcomes with nilotinib.
The safety profile of nilotinib observed in this study was consistent with that reported in earlier studies, with a lower incidence of some of the AEs typically associated with the disease and treatment. The incidence of drug-related thrombocytopenia and anemia were 21.2% and 14.1% compared to 58% and 53%, respectively, reported in the pivotal registration study, and 33.5% and 11.2%, respectively, in the ENACT study.9,17 Interestingly, in the current study, drug-related neutropenia was observed in 8.2% of the patients, whereas in the pivotal registration study and the ENACT study, 53% and 19.2% of the patients reported drug-related neutropenia, respectively. In general, the frequency of grade 3 or 4 AEs was low in the study with 8.2%, 4.7% and 2.4% of patients reporting grade 3 or 4 thrombocytopenia, anemia or neutropenia, respectively. In the ENACT study, 22%, 14% and 3% of patients experienced grade 3 or 4 thrombocytopenia, neutropenia or anemia, respectively.17–19 The incidence of grade 3 or 4 drug-related hematological events was also high in the pivotal registration study, with 30%, 31% and 11% of patients reporting thrombocytopenia, neutropenia or anemia, respectively. 9 The current study reported eight SAEs that were suspected to be drug-related, including one case of poorly controlled diabetes mellitus. In this patient, the SAE was observed at screening, with nilotinib initiated only one week before screening. This patient eventually improved significantly following hospitalization.
During the study, 31 patients (36.5%) discontinued nilotinib, although only 4.7% of the patients discontinued due to AEs, while 16.5% discontinued due to unsatisfactory therapeutic effect. This trend was also observed in other studies evaluating second-line therapies, with a discontinuation rate of 48% in the ENACT study and 61% in the registration study.9,17–19 Notably, the discontinuation rate was 13.3% in the SENSOR study, which only enrolled patients with suboptimal response to imatinib. 20 Among the patients who switched from imatinib to nilotinib due to AEs, 84.2% experienced resolution of their AEs. Minimal cross-intolerance has also been observed earlier in the pivotal registration trial, in which AEs remained unresolved in only 7% of the patients. 21 With a median age of 47 years, the patient population in this study was relatively young for the general CML population, which is also typically seen in the patient population in the Asia-Pacific region.22,23 This might explain the lower incidence of some of the more common AEs seen with the disease and treatment.
Five deaths were reported in this study, of which four occurred in patients with CML-AP at last captured disease status. One death caused by cardiopulmonary failure was suspected to be related to nilotinib. This patient was aged 83 years, with a history of hypertension and cardiovascular comorbidities, including congestive heart failure and coronary atherosclerosis. The patient was in CML-AP at screening, with no improvement in disease state after 1 year of nilotinib therapy, and with CHR being the best response achieved after 54 months of treatment.
Treatment with nilotinib has been shown to be associated with prolongation of the QT interval. 24 Frequent electrocardiogram (ECG) assessments should be performed in patients with CML before initiating nilotinib and during nilotinib therapy. In this study, QTc prolongation was seen in three patients, of whom one experienced grade 3 prolongation but recovered completely with concomitant medication and continued therapy with nilotinib. Only 6% of the patients experienced cardiac and vascular disorders that were suspected to be related to nilotinib, and one instance of hypotension of grade 3–4 severity was reported. There were no instances of peripheral arterial occlusive disease (PAOD). This could be due to the relatively short follow-up period, genetic differences or both. According to previous reports, PAOD appears to increase over time with continuous nilotinib treatment, and has been identified as one of the strongest predictive factors for a higher incidence of CV events in nilotinib-treated patients, 25 although the current study did not collect sufficient data to confirm this.
Overall, 56.8% of the patients in the study achieved MMR by 24 months. As this was an observational study, baseline response rates were not collected and it is possible that some of the patients may have already achieved CHR, CCyR or MMR. Overall, these response outcomes in the study were consistent or better than those seen in the pivotal registration study, in which only approximately 25% of the patients had achieved MMR. 9 However, in the later ENESTcmr study, in which patients crossed over to nilotinib after long-term treatment with imatinib, approximately 40% of the patients achieved undetectable BCR-ABL1 transcripts. 26 Better efficacy of nilotinib than imatinib was also demonstrated in the comparative study of nilotinib with imatinib for newly diagnosed patients with CML, with only 44% of imatinib-treated patients achieving MMR compared to over 65% of patients achieving MMR with both 300 and 400 mg of nilotinib at 24 months. 10 These differences in the response outcomes perhaps reflect better management practices of patients and a greater understanding of the disease and therapies over time.
Patients without CHR at baseline appeared to take longer to achieve response compared to the overall population. Earlier studies have demonstrated better survival and response outcomes for patients who had achieved CHR at baseline compared to those who did not. 9 Similarly, in the ENESTnd study, of the patients who achieved EMR (BCR-ABL1IS ⩽10% at 3 months), 80% in the nilotinib arm and over 58% in the imatinib arm achieved MMR by 2 years. In this study, which assessed nilotinib as a second-line treatment for CML, patients who achieved EMR at 3 months also had better response rates (MMR, 65%; MR4.0, 20%) compared with those who did not achieve EMR (MMR, 14.3%; MR4.0, 0%). Among patients analyzed for EMR at 6 months, the MMR rate was 72% in patients achieving EMR, compared with 30% in patients who did not achieve EMR at 6 months. Similarly, slightly better responses were seen in patients who were intolerant to a previous TKI, as compared to those who were resistant to the previous TKI treatment. Resistance to imatinib is mostly due to the development of BCR-ABL mutations, and the response to second-line therapy is influenced by the sensitivity of nilotinib to BCR-ABL mutations.15,27 Among the imatinib-resistant patients, only 35% of those with BCR-ABL mutations had achieved CCyR, compared to 50% of those without mutations. 9 In this study, few mutations like M351V, F317L, T3151, F359 and E459K were found to be resistant to nilotinib. Mutations G250E, E450G and M244V were sensitive to nilotinib. The responses of mutations F486S and E543A were limited to CHR only.
Limited data were available on EMR, as few patients were eligible for the assessment of BCR-ABL1 at 3 and 6 months due to the observational nature of the study. In addition, response rates immediately after switching to nilotinib could not be estimated as the response rates at baseline were not collected. Although more than 50% of the patients had achieved MMR, most patients did not reach MR4.0 and MR4.5 by the data cutoff at 24 months, which may not be sufficient for these patients to reach MR4.0 or MR4.5. The MR data were also limited due to the observational nature of the study, which considered only those patients who were eligible for the analyses of MR at 3 months.
In summary, the NOVEL study demonstrates that treatment with nilotinib was well-tolerated and was effective in achieving cytogenetic and MRs in patients with CML who were resistant or intolerant to imatinib in the real-world setting in Taiwan. It appears that OS and PFS were significantly influenced by disease status (CML-CP or CML-AP) at screening. However, larger patient numbers are required in each subgroup to draw meaningful conclusions. The safety profile of nilotinib was consistent with earlier reports, and certain factors, such as nonhematological AEs and discontinuations due to AEs, were lower than those reported in earlier studies. Most of the AEs experienced with imatinib that led to intolerance were resolved after switching to nilotinib. These results could be partially explained by a comparatively younger patient population with CML in this region as well as better disease management among clinicians in Taiwan.
Supplemental Material
Supplementary Material
Supplemental material, Supplementary_Figure-1_submission_TAH for Safety and efficacy of nilotinib in routine clinical practice in patients with chronic myeloid leukemia in chronic or accelerated phase with resistance or intolerance to imatinib: results from the NOVEL study by Ching-Yuan Kuo, Po-Nan Wang, Wen-Li Hwang, Cheng-Hwai Tzeng, Li-Yaun Bai, Jih-Luh Tang, Ming-Chih Chang, Sheng-Fung Lin, Tsai-Yun Chen, Yeu-Chin Chen, Tran-Der Tan, Chih-Yi Hsieh, Chinjune Lin, Clinton Lai, Darko Miljkovic and Cheng-Shyong Chang in Therapeutic Advances in Hematology
Supplemental Material
Supplementary Material
Supplemental material, Supplementary_Table_1_submission_TAH for Safety and efficacy of nilotinib in routine clinical practice in patients with chronic myeloid leukemia in chronic or accelerated phase with resistance or intolerance to imatinib: results from the NOVEL study by Ching-Yuan Kuo, Po-Nan Wang, Wen-Li Hwang, Cheng-Hwai Tzeng, Li-Yaun Bai, Jih-Luh Tang, Ming-Chih Chang, Sheng-Fung Lin, Tsai-Yun Chen, Yeu-Chin Chen, Tran-Der Tan, Chih-Yi Hsieh, Chinjune Lin, Clinton Lai, Darko Miljkovic and Cheng-Shyong Chang in Therapeutic Advances in Hematology
Footnotes
Acknowledgements
We thank Amrita Dutta, PhD of Novartis Healthcare Pvt. Ltd. for medical editorial assistance with this manuscript. We would also like to thank Fan-Chen Ku, MD and Yee-Ming Lee, PhD of Novartis (Taiwan) Co. Ltd., for their support during the development of the manuscript and Hsien-Feng Sun of Novartis (Taiwan) Co. Ltd. for monitoring the study. In addition, we also acknowledge the support of the patients and their families.
Authorship
Ching-Yuan Kuo and Cheng-Shyong Chang were involved in study design, data interpretation, writing and reviewing the manuscript and enrolling patients. Jih-Luh Tang was involved in study design, reviewing the manuscript and enrolling patients. Po-Nan Wang, Wen-Li Hwang, Cheng-Hwai Tzeng, Li-Yaun Bai, Ming-Chih Chang, Sheng-Fung Lin, Tsai-Yun Chen, Yeu-Chin Chen and Tran-Der Tan were investigators and enrolled patients. Chih-Yi Hsieh was involved in statistical analysis of study results, preparation of CSR, data interpretation, writing and reviewing the manuscript. Chinjune Lin was involved in study design, data interpretation, writing and reviewing the manuscript. Clinton Lai and Darko Miljkovic were involved in data interpretation, writing and reviewing the manuscript. All authors have approved the manuscript.
Conflict of interest statement
Ching-Yuan Kuo has received honoraria from Novartis, Roche, Celgene, Takeda and Janssen. Cheng-Shyong Chang has received fees for consulting from Novartis, Roche, Takeda, Celgene and Janssen, participated in a speakers’ bureau for Novartis, Janssen and Roche, and received honoraria from Novartis, Roche, Takeda, Celgene, Janssen, Kyrin and MSD. Chinjune Lin, Clinton Lai and Darko Miljkovic are employees of Novartis. Chih-Yi Hsieh was an employee of Novartis from January 2013 to June 2015. All other authors have no conflicts of interest to disclose.
Funding
The study was sponsored by Novartis Pharma AG, Basel.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.