
Abstract
The loss of patents covering many biopharmaceutical/biological agents in the mid 1990s led to the introduction of a new generation of drugs: biosimilars. These new agents, produced by living cells just as the originator drugs, are chemically highly similar to endogenous human proteins; characterized by three-dimensionally complex, high molecular weight compounds. Among the first biosimilars used in haematology–oncology were erythropoietin and granulocyte colony-stimulating factor. After five years of use in clinical practice, the efficacy and safety profile of biosimilars approved by the European Medicines Agency is excellent. Over the next year or two, biosimilar monoclonal antibodies (MoAbs) will become available; the first will be rituximab and trastuzumab. Not only are MoAbs more complex in terms of molecular weight and number of amino acids than the first biosimilars, but they are also anticancer drugs, not merely supportive treatments like their predecessors. This opens up important questions. How are regulatory agencies to assess their clinical efficacy, immunogenicity and safety? Is the neoadjuvant clinical setting the best to evaluate them? What will regulatory agencies decide in terms of switching an originator molecule for a biosimilar or extrapolating efficacy results from one pathology to another? Once biosimilars of rituximab and trastuzumab are approved, several challenging issues will need to be addressed such as how to maintain appropriate pharmacovigilance, how to extrapolate across indications, and issues concerning automatic substitution. There is currently no consensus in any of these areas. This review addresses all these issues: new challenges that the oncology community will face in the near future.
Introduction
The expiry of data protection or patents covering many biopharmaceutical agents in the mid 1990s brought about a new generation of drugs that were copies of the originals, known as originator molecules or just originators, with similar biological activity. They are called ‘biosimilars’ by the European regulatory agency, the European Medicines Agency (EMA), and ‘follow-on-protein products’ by the US regulatory agency, the Food and Drug Administration (FDA). These new agents are chemically very similar to the originators and are characteristically complex, high molecular weight compounds. They are manufactured using either hybridoma or recombinant DNA technology.
Among the first biopharmaceuticals used in haematology–oncology were the haematopoietic red and white blood cell factors [erythropoietins (EPOs) and granulocyte colony-stimulating factor (G-CSF)]. What made these biological medicines unique and different from classical chemotherapeutic agents was the fact that they are produced by living cells [Escherichia coli and Chinese hamster ovary (CHO) cells] that generate a mixture of related molecules, which makes them difficult to characterize. Chemical medicines, in contrast, are made by chemical processes, have a relatively simple and well-defined structure, and are easy to characterize. Each of the new biopharmaceutical agents can be extremely similar to others and to the reference molecule (originator), but they are not identical, no matter how similar are.
Over the past five years, biosimilars have been introduced in many parts of the world and in particular in Europe. It appears that, after a long debate, biosimilars will enter the US market next year. In addition, a new landmark will emerge in the field, a new generation of biopharmaceuticals will be born: biosimilars known as monoclonal antibodies (MoAbs). The complexity of these drugs represents a new challenge; the structure of an immunoglobulin molecule far exceeds that of previous biosimilars.
This review attempts to describe the state of the art and what we have learnt from the first wave of biosimilars (EPOs and G-CSF). We also hope to give some idea of what to expect from this new generation of highly complex molecules, MoAbs. We consider their production, complexity, clinical development and safety, as well as the new challenges that this field will face in the near future.
The clinical development of biosimilars can be presented as falling into three levels (Professor C. Twelves, personal communication) (Table 1). At the first level, we have the first biosimilars: hormones (growth hormone) and haematopoietic growth factors (filgrastim and EPO). The biosimilars at this level present a fast response and have excellent efficacy and safety profiles. At level 2 there are agents of the type of tumour necrosis factor inhibitors (TNFi). They present a fast to intermediate response; in terms of efficacy and safety they appear to be excellent, but there are not enough data to evaluate them rigorously. At level 3, there are proteins with a more remote, long-term response. The agents at this level will include targeted therapies such as MoAbs (trastuzumab, rituximab). The question of how to evaluate these new biosimilar agents that are ready to be commercialized represents a major challenge. At this level, many are anticancer therapies with a wide variability in terms of efficacy, depending on patient characteristics. Testing such new drugs in a neoadjuvant setting may be an elegant method to analyse their efficacy and safety.
Biosimilars classified by molecular complexity and time to response (Professor C. Twelves, personal communication).

G-CSF, granulocyte colony-stimulating factor; MoAb, monoclonal antibody; N/A, not available; TNF-α, tumour necrosis factor-α.
Haematopoietic growth factor biosimilars
EPOs and filgrastim (G-CSF) were the first biosimilars to be produced for use in haematology–oncology. Pegfilgrastim biosimilars were ready to be authorized by the end of 2014.
G-CSF
G-CSF is a 20 kDa glycoprotein composed of a single polypeptide chain of 174 or 177 amino acids. It is glycosylated at the threonine residue 133. Native G-CSF is located on chromosome 17 and due to differential splicing encodes two protein products: isoform A, of 177 amino acids; and isoform B, of 174 amino acids. The three additional residues in isoform A (Val–Ser–Gln) are inserted after Leu35. The 174 amino acid form of the molecule is associated with greater biological activity and stability than the longer isoform. This difference forms the basis for commercial pharmaceutical G-CSF products, including Neupogen® (filgrastim). G-CSF is the haematopoietic growth factor that is most important for the recovery of neutrophils: it stimulates the proliferation of neutropenic progenitor cells, participates in their differentiation to granulocytes and functionally activates mature neutrophils [Aapro et al. 2010].
Mechanism of action
Human G-CSF is a lineage-specific CSF produced by fibroblasts, monocytes and endothelial cells. G-CSF regulates the production of neutrophils within bone marrow (BM) and induces neutrophil progenitor proliferation, differentiation and specific cell functional activation (including enhanced phagocytic activity such as priming of the cellular metabolism-associated burst and antibody-dependent killing). G-CSF is not species-specific and has been shown to have minimal or no direct in vivo or in vitro effects on the production of haematopoietic cell types other than the neutrophil lineage. It acts by binding to a specific transmembrane receptor (G-CSF R), a member of the class I cytokine receptor family expressed in various haematopoietic cells such as stem cells, multipotent progenitors, myeloid-committed progenitors, monocytes and neutrophils. The receptor forms homo-oligomeric complexes upon ligand binding. The same receptor–ligand mechanism operates in the mobilization of mature neutrophils and their recruitment into the circulating neutrophil pool, and in increasing granulopoiesis.
Indications
Filgrastim (Neupogen®) was one of the first biopharmaceutical human recombinant G-CSF products to be commercialized and it is the reference drug for all G-CSF biosimilars.
The regulatory agencies have approved its use for the following treatments of clinical conditions [Aapro et al. 2010].
Reduction of the duration of neutropenia and the incidence of febrile neutropenia in patients treated with cytotoxic chemotherapy for malignancy (with the exception of myeloid malignancies); and reduction in the duration of neutropenia in patients undergoing high-dose chemotherapy followed by BM transplantation. The efficacy and safety of filgrastim are similar in adults and children receiving cytotoxic chemotherapy.
Mobilization of peripheral blood progenitor cells.
In patients (children or adults) with idiopathic or severe congenital cyclic neutropenia with an absolute neutrophil count (ANC) of ⩽0.5 × 109/l and a history of severe or recurrent infections, long-term administration is indicated to increase neutrophil counts and to reduce the incidence and duration of infection-related events.
Treatment of persistent neutropenia (ANC ⩽ 1.0 × 109/l) in patients with advanced human immunodeficiency virus (HIV) infection, to reduce the risk of infections when other therapeutic options are inappropriate.
Filgrastim (G-CSF) biosimilars
Several G-CSF biosimilars have been EMA approved over the last few years: biograstim/Filgrastim ratiopharm/ratiograstim/tevagrastim (XM02); Zarzio® and Nivestim® (Table 2).
Filgrastim biosimilars authorized in Europe.
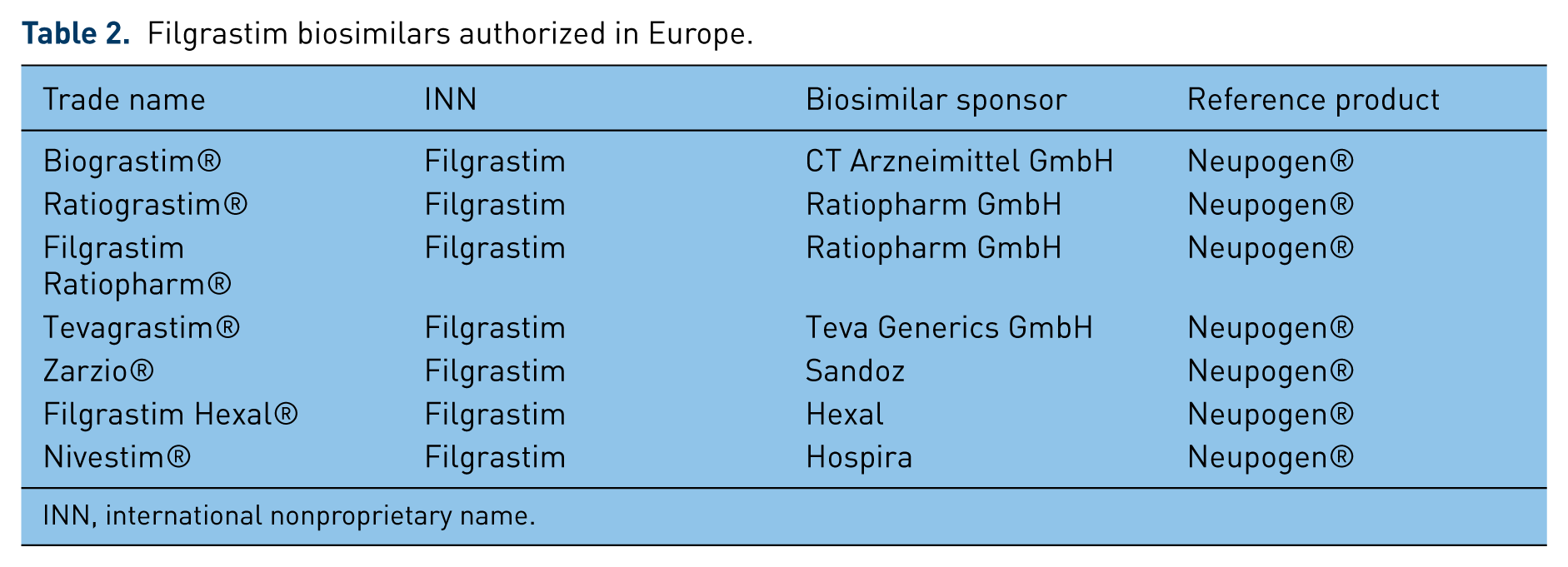
INN, international nonproprietary name.
Biograstim/Tevagrastim (XM02)/Ratiograstim
Active substance
The active substance of this G-CSF- biosimilar (XM02) is a recombinant human G-CSF produced in E. coli, yielding a nonglycosylated protein with an N-terminal methionyl extension; its international nonproprietary name (INN) is filgrastim [EMA, 2008a, 2008b]. The protein is known to be expressed in inclusion bodies followed by the renaturation of protein and several purification chromatographic steps. The protein is a recombinant form of the 174 amino acid isoform which contains an additional N-terminal methionine residue that is not found in the native human protein. Naturally occurring G-CSF is glycosylated at the threonine residue 133, a modification which is absent from the active XM02 compound as an E. coli expression product (bacteria do not produce glycosylated products, unlike mammalian cells).
Presentation
This G-CSF biosimilar is supplied in prefilled syringes containing 0.5 ml (lower strength) or 0.8 ml (higher strength) of sterile, preservative-free solution for injection, consisting of 30 or 48 million units (MU) (corresponding to 300 μg and 480 μg, respectively) of the XM02 active substance together with acidic sodium acetate buffer, polysorbate, sorbitol and water for injection. This formulation is very similar to Neupogen® and only slight differences exist in the concentration of the polysorbate 80 and the pH value. It is administered via subcutaneous (SC) or intravenous (IV) injection, normally at a dose of 1–0 μg/kg/day, depending on the indication.
Pharmacodynamic (PD) and pharmacokinetic (PK) studies
This biosimilar underwent two phase I studies comparing it with Neupogen®. The studies were performed on healthy volunteers. The mean serum concentration was 5 and 10 μg/kg of the active compound of XM02, similar to that of Neupogen®. One single injection of the active XM02 agent produced the same CD34+ cell count peak at 72 hours, which returned to baseline levels after 336 hours; exactly as for the originator, Neupogen®.
Clinical efficacy
A pivotal study to assess clinical efficacy was carried out on patients with breast cancer who were treated with the reference product (Neupogen®) for up to four cycles of chemotherapy. The clinical efficacy of XM02 was evaluated and considered to be comparable with Neupogen®. There were no immunogenicity findings of clinical relevance which had any major consequences for efficacy and safety in the three studies. Patient blood samples were studied for the presence of neutralizing antibodies in plasma; none were detected.
Safety
Strict evaluations of XM02 for safety included analysis of five clinical studies: two phase I studies performed with healthy volunteers; and three studies in cancer patients: breast and lung cancer and non-Hodgkin’s lymphoma. The lung cancer and non-Hodgkin’s lymphoma studies were designed primarily to assess the safety of XM02. In a pooled analysis of the three studies, the incidence of several treatment-emergent adverse events (TEAEs) (in cycle 1: alopecia, neutropenia, asthenia, diarrhoea, bone pain and abdominal pain) was statistically higher in the Neupogen® only group than in the XM02 only group. However, these differences have proven to be of no clinical relevance.
ZARZIO®
Introduction
Human G-CSF (filgrastim) is produced by rDNA technology in bacteria (E. coli) from the full-length human sequence for N-(
Presentation
Currently, two presentations of the factor are available: 30 MU (300 μg/0.5 ml) in a prefilled syringe; and 48 MU (480 μg/0.5 ml), also in a prefilled syringe.
PD studies
Four [EMA, 2008c] studies of PK/PD were performed on healthy volunteers. PD activity was based primarily on ANC peak response and ANC exposure, that is, the whole area under the curve (AUC) over 10 days. The overall results support the comparability of the biosimilar and the reference product with respect to their PD, since the ANC curves can be superimposed, regardless of the route and the dose used. The CD34+ cell count after repeated dosing (secondary PD endpoint) demonstrated a similar time profile for filgrastim and Neupogen®, and AUEC0-216h .
PK studies
Serum levels of free G-CSF were found to be lower after administration of filgrastim than after Neupogen®; the difference appeared consistent across the routes and doses, and was statistically significant. Bioequivalence was observed between the two products in terms of their elimination halflives. It is known that the apparent difference in bioavailability may be overestimated due to the nonlinear saturable PK of rhG-CSF, a large part of which is eliminated through binding to its target cells: myeloid progenitors and neutrophils. A very plausible explanation of the difference in elimination at different doses is that receptor-mediated clearance (which is saturable) is predominant at lower doses, while renal clearance becomes more important at higher doses.
Efficacy
A study in healthy volunteers to assess the efficacy of the filgrastim biosimilar (Zarzio®) using as an endpoint the ANC and CD34+ cell count was considered acceptable by the Committee for Medicinal Products for Human Use (CHMP) at the EMA. Moreover, the extrapolation to all indications of the originator is acceptable, since the mechanism of action is the same: direct stimulation of BM cells through one specific type of surface receptor.
Safety
The comparison of the safety profile of this biosimilar and that of the originator was based on studies in healthy volunteers. A total of 4 studies recruited 146 healthy volunteers. The adverse drug reaction (ADRs) associated with filgrastim were consistent with those published in normal healthy volunteers reported for Neupogen®. Overall, these data supported the comparability of the two products. The small single-arm trial in cancer patients submitted to the regulatory agencies allows, to a certain extent, unexpected safety events to be ruled out and suggests low immunogenicity of the product (Zarzio®). Additional long-term safety and immunogenicity data are being collected postmarketing.
Nivestim®
Introduction
Nivestim® is a recombinant r-metHuG-CSF protein consisting of 175 amino acids that is produced, as other biosimilars are, in the bacteria E. coli. It has a molecular weight of 18,800 Daltons [EMA, 2010]. Unlike the human protein, Nivestim® is unglycosylated and contains an N-terminal methionine. Filgrastim, the active substance in Nivestim®, is itself also a recombinant human G-CSF produced in E. coli as a nonglycosylated protein containing an N-terminal methionyl extension. It functions as the human factor does: it stimulates the proliferation, differentiation and activation of late progenitor cells of the granulocyte lineage, as well as enhancing the function of mature neutrophils [EMA, 2010].
Presentation
Nivestim® is available in solution for injection or infusion. It comes in three strengths: 120 μg/0.2 ml, 300 μg/0.5 ml and 480 μg/0.5 ml. The qualitative and quantitative composition of Nivestim® and Neupogen® has been demonstrated to be the same, with the exception of the 120 μg/0.2 ml presentation, which is not marketed by Amgen®. Nevertheless, this presentation only differs in fill volume from the 300 μg/0.5 ml presentation.
PD and PK studies
Two PD/PK studies have been performed in healthy volunteers [EMA, 2008a]. Two phase I trials were carried out as PK studies and to demonstrate the biosimilarity between Nivestim® and Neupogen®, the reference product/originator. The primary endpoint ANC AUC was also equivalent to Neupogen® for both IV and SC administration. The results of these studies show that the mean ANCmax, ANCmin and CD34+ cell count were equivalent for individuals receiving either Nivestim® or Neupogen®; while ANC Tmax occurred slightly sooner in those receiving treatment with Nivestim® (7.845 hours) than in those treated with the originator Neupogen® (9.448 hours).
Efficacy
The efficacy of Nivestim® is similar to that of Neupogen®. The clinical development of Nivestim® to demonstrate biosimilarity between Nivestim® and Neupogen® included one phase III study, which was performed in women with breast cancer who received G-CSF prophylaxis in addition to a normal chemotherapy regimen. Therapeutic equivalence was observed between the two factors in terms of efficacy as analysed by mean ANC nadir and with regard to time to ANC recovery.
Safety
This is similar to that of Neupogen®. The Nivestim® study group appeared to contain a greater proportion of patients with severe neutropenia than the Neupogen® study group in cycle 1 and cycle 2. Furthermore, there seemed to be a lower proportion of subjects treated with Nivestim® who developed severe neutropenia in cycle 3 than those treated with Neupogen®, and the duration of severe neutropenia (DSN) seemed to be longer in the Nivestim® study group. However, these findings were not statistically significant. The most common adverse reaction was bone pain, with a higher incidence in the Nivestim® group (Nivestim®: n = 26, 14.2%; Neupogen®: n = 9, 9.5%). All of these events were considered mild or moderate in nature, except for one subject on Neupogen® who manifested severe pain in the extremities [EMA, 2010].
Lipegfilgrastim (XM22)
This new G-CSF biosimilar was ready to be commercialized by the end of 2014 or early 2015. It is a biosimilar of the original pegfilgrastim (Neulasta®) and it is produced by Teva Pharmaceuticals®. In a phase II, dose-ranging study to determine the optimal fixed dose of lipegfilgrastim compared with that of pegfilgrastim (6 mg), 208 chemotherapy-naïve patients with breast cancer were randomized to receive the biosimilar at a dose of 3.0, 4.5 or 6.0 mg, or pegfilgrastim at 6.0 mg [Buchner et al. 2014]. The pharmaceuticals were administered on day 2 of each chemotherapy cycle (doxorubicin/docetaxel on day 1 for four 3-week cycles). The primary outcome was the DSN in cycle 1. Patients treated with lipegfilgrastim showed a shorter DSN in cycle 1 at higher doses. The mean DSN was 0.76 days in the lipegfilgrastim 6.0 mg group and 0.87 days in the pegfilgrastim 6.0 mg group. However, no significant differences were observed among the different treatment groups. Interestingly, treatment with lipegfilgrastim 6.0 mg was associated at nadir counts with a higher ANC, shorter ANC recovery time, and an excellent safety and tolerability profile, similar to pegfilgrastim. The authors state that the study demonstrated that lipegfilgrastim 6.0 mg is the optimal dose (exactly as for Neulasta®) for patients with breast cancer and provides neutrophil support that is at least equivalent to that provided by pegfilgrastim (6.0 mg), the originator [Buchner et al. 2014].
Summary of filgrastim biosimilars
G-CSF biosimilars (biograstim/filgrastim ratiopharm/ratiograstim/tevagrastim (XM02); Zarzio® and Nivestim®) are manufactured in facilities with state-of-the-art technology. All the products have passed the regulatory requirements for approval [Schellekens, 2009], mainly a phase I and a phase III trial with the consequent PD/PK evaluations and studies of efficacy and safety. However, there are still some concerns regarding the long-term evaluation of these products; particularly, the limited experience with these products in terms of efficacy, safety and immunogenicity at the time of approval. For this same reason, pharmacovigilance should be rigorous and is important as a public health concern. Guidelines will allow the extrapolation of data to additional indications, for example, paediatric indication and peripheral blood progenitor cells (PBPC) in healthy donors. Much further work is necessary in terms of clarification with regard to substituting a biosimilar G-CSF for the originator. Finally, information must be provided to physicians, pharmacists and patients to allow for proper decision making. Ultimately, only clinical trials and effective postmarketing pharmacovigilance will provide definitive evidence that a biosimilar is comparable with the reference product in term of efficacy and safety [Roger, 2010].
Epoetin biosimilars
Three epoetin biosimilars are authorized by the EMA: HX575; XM01 (this is in fact an originator, according to its clinical development); and SB309 (Table 3). The names of these three agents change as they are commercialized. The manufacturing process has to follow a rigid quality control protocol in order to ensure efficacy and safety profiles similar to those of the originator molecule.
Epoetin biosimilars authorized in Europe.

INN: international nonproprietary name.
The active molecule or pharmaceutical substance is identified by its INN. This name is important both for physicians and for pharmacists when dealing with substitution issues and for postmarketing follow up. An active substance with its own INN can be commercialized by different companies, in which case they use its commercial registered trademark name. In Europe, a company can choose a name for a biosimilar that can be identical to or different from the original. Thus, HX575 has been registered as epoetin α, even though its carbohydrate differs from the originator Eprex®/Erypo®. HX575 presents high levels of mannose and low levels of both N-glycolyl-neuraminic acid and diacetylated neuraminic acid [EMA, 2007a]. This can generate confusion as in the case of HX575, which has been registered under three different names: Binocrit® (Sandoz), Epoetin alfa-Hexal® (Hexal Biotech) and Abseamed® (Medice Arzneimittel Putter). Another biosimilar, SB309, has been registered under the name epoetin theta and commercialized under the names: Silapo® (Stada) and Retacrit® (Hospira). This agent has fewer O-glycanes and lower levels of both N-glycolyl neuraminic acid and O-acetyl neuraminic acid than the reference product [EMA, 2007b]. The third of the epoetin biosimilars is epoetin theta. Although, this agent is for all practical uses a biosimilar, in reality it belongs to the originator family since all its clinical development has been exactly the same as for the originator. It has been commercialized under the names Eporatio® (Ratiopharm-TEVA) and Ratioepo® (Ratiopharm-TEVA). With regards to N-glycosylation, there are no qualitative differences but there are quantitative ones. XM01 contains more N-linked tetrasialioside structures (70.1%), which are essential for a long halflife in vivo, trisialo- (17.0%) and disialo- (7.6%). Furthermore, XM01 is completely glycosylated.
HX575
HX575 (INN: epoetin alfa) has been registered under the name Binocrit® (Sandoz). It has been produced in CHO cells. It has been compared with the original Eprex®/Erypo® in preclinical and clinical studies. The agent has been characterized according to the primary structure via matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) analysis covering 99% of the molecular sequence. Secondary and tertiary structure analysis shows correct folding of the molecules as well as excellent consistency between different batches. The N- and O-glycane structures have been studied quantitatively as well as qualitatively, revealing close similarity with the originator. The studies have detected high levels of glycanes in manose-6-phosphatase (HM6P) in some batches utilized in clinical trials. The levels of HM6P detected in HX575 did not affect the efficacy or the safety of the compound. HX575 had lower values of N-glycolyl-neuraminic acid and diacetylated neuraminic acid than the originator, Eprex®/Erypo® [EMA, 2007b].
Preclinical studies
The biological activity of HX575 was determined both in vitro and in vivo. Five nonclinical, pharmacological and PK studies were performed on mice, dogs and rabbits prior to registry. All these PK/PD studies showed great similarity with the originator.
Clinical studies
Five pharmacological studies (PK/PD) were performed on healthy male volunteers who received one or multiple doses of the compound by IV or SC administration [EMA, 2007b; Sorgel et al. 2009]. Four of the studies used either Erypo® (epoetin alfa) or NeoRecormon® (epoetin beta) as the originator molecule. In addition, two phase III, multicentre, double-blind randomized studies were performed. One compared the efficacy and safety of HX575 and Erypo® via IV administration to patients with anaemia secondary to chronic renal insufficiency (CRI), with 314 patients in the HX575 arm and 164 in the Erypo® arm. A second study evaluated the efficacy and safety of HX575 in 114 patients with chemotherapy-induced anaemia [Kramer, 2008; Schellekens and Moors, 2010].
Based on the information facilitated by the manufacturer of HX575 to the regulatory agency, the CHMP review recommended the use of HX575 in: (a) anaemia associated with CRI in children and in adult patients undergoing haemodialysis as well as in adults on peritoneal dialysis; (b) patients with CRI with symptomatic anaemia but not undergoing dialysis; (c) anaemia secondary to chemotherapy for solid tumours, malignant lymphoma and multiple myeloma; and (d) reducing the risk of allogeneic transfusions in adults before elective orthopaedic surgery.
XM01
XM01 [INN: epoetin theta; Ratioepo® (Ratiopharm-TEVA), Eporatio® (Ratiopharm-TEVA)] is a glycoprotein of 165 amino acids produced using recombinant DNA technology in CHO cells. Its sequence of amino acids is identical to that of human EPO (HuEPO) and to recombinant HuEPO (alfa and beta; with the exception of darbepoetin alfa, since that molecule has five amino acids that are different from those of the original). The only difference with the original EPO is the glycosylation pattern. It is important to note that XM01 has been developed as a ‘new active substance’ under Article 8(3) of Directive 2001/83/EC, and as a consequence, it is not a biosimilar since all its clinical development has been the same as for the original. During its clinical development, XM01 has been compared with Eprex®/Erypo®, Epoetin alfa-Hexal® (epoetin alfa) and NeoRecormon® (epoetin beta), showing a high degree of similarity with other EPOs. Chemical analysis of XM01 has demonstrated that the content of oligosaccharides is the same as in the other authorized and commercialized EPOs. However, XM01 presents some differences in the number of glycanes it contains.
XM01’s liquid formulation contains a phosphate Tris buffer and polysorbate 20. MALDI-TOF analysis of O-glycosylation demonstrated 100% homology with NeoRecormon®. The composition of the tetrasialo was shown to be 70.15% N-glycosylated and that of Erypo® was 62.1%; while Epoetin alfa-Hexal® was 64.7% and NeoRecormon® 57.7%. The XM01 molecule presents higher degrees of sialization in all EPOs. All studies confirm that the XM01 sequence of amino acids is exactly the same as that of the human EPO and epoetin alfa and beta.
In summary, in preclinical studies: characterization of XM01 and the proteins of its originator shows a close similarity between them; and the different chemical structures between XM01 and the other known EPOs are minimal, quantitative and do not induce any unwanted biological side effects.
Clinical studies
During its clinical development there have been 15 registered studies: 6 phase I, PK and safety trials; 2 phase II trials; and 7 phase III trials. In a pivotal study in patients with chronic kidney disease (CKD) in haemodialysis, complete similarity in terms of efficacy and safety of IV epoetin theta were observed in the maintenance phase compared with epoetin beta [Gertz et al. 2010]. In another study with XM01 in patients treated with platinum (n = 150), a response rate of 20.3% was demonstrated for the placebo arm and 65.8% in the XM01 arm [for haematological responses, haemoglobin (Hb) ⩾ 2 g/dl] and of 50% and 90.8% for those patients considered to be partial responders (Hb ⩾ 1 g/dl) after 4 weeks. For those patients treated with non-platinum in the XM01-22 study (n = 186), the response rate was 25.3% for the placebo arm and 72.6% for the XM01 arm. For partial responders, the response rate was 61.5% for the control arm and 82.1% for the experimental arm [Tjulandin et al. 2010, 2011].
In patients with chemotherapy-induced anaemia, XM01 is clinically comparable with NeoRecormon®, but at a lower initial dose and weekly maintenance [20,000 international units (IU)/week).
SB309
The INN of the agent SB309 is epoetin theta and its commercial names are Retracrit® and Silapo®. This agent was approved by the European Commission in 2007 as a biosimilar with epoetin alfa (Eprex®/Erypo®) as the originator. It was approved for anaemia associated with CKD and for anaemia induced by chemotherapy.
Clinical studies
The SC half-life of this agent is 24 hours, exactly the same as for NeoRecormon®, and the discontinuation of the cold chain (up to 25°C) is 3 days, the same as for the originators Eprex® and NeoRecormon®. Silapo®, via IV administration was compared with the originator (Eprex®/Erypo®) in 2 pivotal studies involving a total of 922 patients with anaemia associated with CKD undergoing haemodialysis. The first of the studies analysed the correction of anaemia in 609 patients over a period of 24 weeks. In the second, 313 patients were recruited and SB309 was compared with the originator in the maintenance of haemoglobin. All the patients in the second study had been treated with (Eprex®/Erypo®) for at least 3 months before they were either changed to Silapo® or left with the same originator for an extra 12 weeks. After that, the groups interchanged medications for another 12 weeks. Efficacy was considered as the levels of Hb reached during the study and the total dose of epoetin administered to each patient.
In the first trial, aimed at the correction of Hb, the levels reached were of 11.6 g/dl over the 4 week period, starting from basal levels of around 8 g/dl. In the second study, aimed at the maintenance of the Hb levels, it was observed that Silapo® maintained Hb levels of around 11.4 g/dl, similar to the levels in patients who were receiving the originator (Eprex®/Erypo®). It was also noted that the total levels of EPO received by the patients was similar in both groups.
With regard to the use of SC Silapo®, two studies were presented to the European Commission. The first studied 261 patients with cancer and the second 462 patients with anaemia secondary to CKD. The results in these cases were also very similar to those published in the literature for the original EPOs [EMA, 2009].
A pivotal phase III trial with Retracrit® (SB3099) studied the correction of anaemia induced by chemotherapy in 216 patients with solid tumour or nonmyeloid haematological malignancies [Tzekova et al. 2009]. The biosimilar demonstrated a significant increment in Hb of 1.8 g/dl over the 12 weeks of the study. A total of 81.5% of the patients reached an increment of ⩾ 1 g/dl in the first 8 weeks of treatment, while 70.8% reached levels of Hb of ⩾ 2 g/dl. These results are very similar to those for the original EPOs. In addition, it was demonstrated that 81% of the patients had not received any EPO during the time of the study. Furthermore, patient quality of life improved. Safety data were very similar to those for epoetin alfa and for epoetin beta [Cazzola et al. 2003]. Around 4.2% of the patients presented some thromboembolic event in the first 12 weeks. These results are similar to the 4.5% published in a meta-analysis of 35 studies comprising a total of 6,769 patients [Bohlius et al. 2006].
Pure red cell aplasia (PRCA)
One of the most serious potential complications when agents similar to the endogenous human substances (EPO, G-CSF) are given to humans is the possibility of originating antibody formation. In other words, the possibility that the new agent, in this case a biosimilar, behaves like an immunogen and therefore originates an immune response. In the case of recombinant EPOs (r-EPOs), there would be destruction of the endogenous EPO as well as of the EPO biosimilar, with grave consequence, since the BM would not be capable of producing red blood cells; this leads to the clinical condition known as pure red cell aplasia (PRCA). Clinically, the patient develops a rather rapid and severe normochromic, normocytic anaemia. The anaemia can lead to a drop in Hb of 1 g/l/day, associated with severe reticulopenia (<10,000 per μl) and a lack of erythroid precursors in the BM. Clinical confirmation is the identification of neutralizing anti-EPO antibodies in the plasma of the patient. This situation occurred in 1998 and 2003 with the original epoetin alfa [Casadevall et al. 2002]. More than 200 cases were identified worldwide. Interestingly, PRCA was only detected in patients with CKD who received the agent via IV administration and not among cancer patients. The manufacturer eliminated the traces of albumin present in the original, substituting it with polysorbate 80 and glycine to avoid the risk of prion transmission. At the same time, the company introduced a prefilled syringe with a rubber cup. When the runner was changed to Teflon™, the incidence of PRCA decreased and reached zero reports.
The question remains as to why PRCA only occurred in CKD patients. One plausible explanation is that cancer patients receiving chemotherapy are already immunocompromised, either by the cancer or most likely by the chemotherapy itself. This incident represents an important lesson in the field of biosimilars, even though the events happened with the originator. The lesson is that biosimilars are extremely complex molecules, with secondary and tertiary structures that are highly unstable and vulnerable to changes in temperature or to becoming immunogenic with minimal chemical or conformational changes in the molecule during production. In more recent times, two potential cases using biosimilars have been identified. In the study named SWEEP, the agent HX575 was given via SC administration to patients with anaemia associated with CKD [Mikhail and Farouk, 2013; Abraham and MacDonald, 2012]. Two cases were reported, one confirmed by the presence of neutralizing anti-EPO antibodies and BM analysis. The second presented neutralizing anti-EPO antibodies, although in this case the BM analysis was not performed due to the death of the patient from cardiac failure. Interestingly, the agent HX575 presents fewer aggregates, a parameter of excellent production, than the originator Erypo®. Fortunately for the field, the lack of more cases suggests that both cases were caused by external factors, such as the cold chain, and were not due to intrinsic defects of the biosimilar.
There have been other cases of PRCA, but they were mainly detected in Thailand. In this case, the so-called biosimilars are not biosimilar at all but pure ‘copies’ of the originator. The agents have not been approved by the EMA and the quality control of those ‘copies’ is, at the very least, questionable.
Conclusion
Three epoetin biosimilars have been commercialized: two genuine biosimilars, Binocrit® and Retracrit®, and one original epoetin theta (Ratioepo®). All three have been registered by the EMA which gives a guarantee of efficacy and safety after having successfully passed all the strict quality controls of the European agency. The issue of automatic substitution of an originator for a biosimilar remains. There is no consensus among the different countries that commercialize biosimilars. It is important that physicians participate in discussions since it is vital that the prescribing physician is in control of what the patient receives because, among other reasons, because the prescribing physician is ultimately responsible for the care of the patient. Years of postmarketing follow up will be necessary to detect any potential problem [Pritchard, 2011].
MoAB biosimilars
MoAbs, Paul Ehrlich’s ‘magic bullets’, were first developed in 1975 using the hybridoma technique. Unfortunately, the first MoAbs to be made were immunogenic in humans, since the cell of origin was murine. Later and thanks to advances in antibody engineering, it became possible to develop chimeric, humanized and fully humanized MoAbs for human use [Weiner et al. 2010]. There has been a real explosion of MoAbs successfully reaching the clinic (rituximab, trastuzumab, cetuximab, panitumumab, bevacizumab, adalimumab, infliximab) for the treatment of different human conditions.
Because of the development of copies of the original biopharmaceuticals that are available in many countries around the world, maintaining postmarketing quality control and additional long-term safety data, together with the collection of immunogenicity data, are of paramount importance. This is a major concern, since there are no consistent worldwide requirements for the registration of so-called biosimilars, and not all of them have undergone the same strict regulation as those approved by the EMA. According to the EU, a biosimilar agent is a copy of an already authorized biological product, the originator or reference product, with demonstrated similarity in physicochemical characteristics, efficacy and safety. In contrast, FDA guidelines to define the data requirements for biosimilars and interchangeable biosimilars are still being drafted, and after much debate, it is likely that biosimilars will enter the US market in 2015.
A MoAb is a very complex molecule (MW: 150 kDa) when compared with an antibiotic or aspirin (MW: 0.18 kDa) or to a simple biologic such as filgrastim (19 kDa) or even the more complex EPO (34 kDa) (Figure 1). Due to the molecular complexity, current analytical tools may not be sufficient to detect minor differences such as post-translational modifications or protein folding. This is a serious issue since minor modifications to the structure of the molecule can lead to different bioactivity and immunogenicity [Mellstedt, 2013; Rak Tkaczuk et al. 2014]. For the past five years, we have been using simple biologic biosimilars such as EPO or filgrastim, agents used for supportive care, mostly in cancer patients. Biosimilar MoAbs are anticancer agents, targeted agents; here both efficacy and safety are of paramount importance for the final outcomes of our cancer treatments.

Molecular complexity of biologics compared with that of small molecules.
Although not used by oncologists, the first biosimilar MoAbs were approved in 2013 by the EMA’s CHMP. The approval was for two biosimilar infliximab products: Celltrion’s Remsima® and Hospira’s Inflectra®. The first MoAbs for haematology–oncology, whose data protection or patents will expire over the next couple of years, are rituximab and trastuzumab.
Rituximab
Rituximab is a MoAb against CD20. It is used to treat B-cell lymphoproliferative malignancies and B-cell mediated autoimmune diseases. Several companies have already produced or are developing rituximab biosimilars for a good reason; rituximab has the largest market of any therapeutic MoAb worldwide [Qureshi et al. 2013; Vital et al. 2013]. In this review, we discuss three rituximab biosimilars: GP2013 (Sandoz®), Reditux® (Dr Reddy’s®) and PF-05280586 (Pfizer).
GP2013
The agent GP2013 is a rituximab biosimilar [Visser et al. 2013]. To compare it with its originator, a series of physicochemical and functional tests were performed which characterized this agent as a biosimilar. Primary and higher-order protein structures, peptide mapping, and both charge and amino acid modifications were analysed. Glycans were identified and quantified as well as glycan site occupancy. Biological characterization included a series of bioassays [in vitro target binding, antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC) and apoptosis] and fragment, crystallisable (Fc) receptor binding assays. GP2013 was shown to be physicochemically highly similar to the originator, rituximab, at the level of primary and higher-order structure, post-translational modifications and size variants. An extensive functional characterization package indicated that GP2013 has the same biological properties as the originator, rituximab [Visser et al. 2013].
In another preclinical study, post-translational modifications to and the bioactivities of GP2013 were studied and monitored, then compared with the originator, rituximab, to establish pharmacological profile similarity.
Pharmacological comparability between the biosimilar GP2013 and rituximab were confirmed in preclinical studies using a clinical-scale drug product. Similar in vitro ADCC potency was demonstrated when its dose–response activity against 2 lymphoma cell lines using human natural killer (NK) cells was compared with that of the originator. In vivo efficacy was demonstrated in two xenograft murine models at sensitive subtherapeutic doses. PK and PD results (CD20 cell depletion) were comparable in cynomolgus monkeys. These preclinical comparability data confirm that the biosimilar GP2013 and the originator rituximab are pharmacologically similar [Da Silva et al. 2014].
Reditux®
Another rituximab biosimilar, Reditux®, was approved in India in 2007. Recently, a retrospective audit was conducted to compare the efficacy and safety of the originator, rituximab (Mabthera®), and Reditux® [Roy et al. 2013]. The treatment charts of 223 adult diffuse large B-cell lymphoma patients were reviewed. Patients had received cyclophosphamide, doxorubicin, vincristine and prednisolone together with rituximab chemotherapy. Data on tumour recurrence, survival and toxicity experienced during treatment were obtained from the charts. Only patients who had received at least four cycles of the same brand were analysed in the survival analysis.
Of the 223 patients evaluated, 72 received Reditux® and 101 received Mabthera®. There were no differences in the infusion reaction rates, grades 3 and 4 neutropenia, or oral mucositis between the biosimilar and the originator. Complete remission (CR) rates were similar with Reditux® and Mabthera® (82% and 75%, respectively; p = 0.294). The progression-free survival (PFS) rates at 5 years were 81% for Reditux® and 72% for Mabthera® (p = 0.382). The overall survival (OS) rates at 5 years were comparable in the two groups (76% in Reditux® and 66% in Mabthera®; p = 0.264). The authors concluded that no significant differences in terms of toxicity, tumour response rates, PFS and OS between the two were observed [Roy et al. 2013].
PF-05280586
Preclinical studies were performed to compare the proposed biosimilar PF-05280586 and rituximab-EU (MabThera®) [Ryan et al. 2014]. Results from side-by-side studies, peptide maps and complement-dependent cytotoxicity assay were similar for the biosimilar and the originator. Cynomolgus monkeys were administered PF-05280586 or rituximab-EU as a single dose of 0, 2, 10 or 20 mg/kg on day 1 and observed for 3 months (single-dose study), or as 5 weekly injections of 0 or 20 mg/kg. The animals were necropsied on day 30, the day after the 5th dose, or on day 121 (in the repeat-dose study). The PK/PD profiles of both molecules were similar. A marked reduction of peripheral blood B cells 4 days after dosing was followed by near or complete recovery (single-dose study) or partial recovery (repeat-dose study). In the single-dose study, anti-drug antibodies (ADA) were identified by day 29 in all animals administered PF-05280586 or rituximab-EU, and they persisted through day 85, the last day they were tested for. In the repeat-dose study, ADA were detected on day 121 in 50% of the animals administered rituximab-EU or PF-05280586. Both molecules were well tolerated at all doses. In all the preclinical endpoints evaluated, the two agents, PF-05280586 and rituximab-EU, demonstrated close similarity [Ryan et al. 2014].
Trastuzumab
Trastuzumab (Herceptin®) is a humanized MoAb that binds to the human epidermal growth factor receptor type 2 (HER2) protein. It is the treatment of choice for women with breast cancer who overexpress HER2. In the adjuvant and metastatic setting, trastuzumab has been shown to improve disease-free survival (DFS) and OS. More recently, trastuzumab has been used in a neoadjuvant setting with excellent results. Contrary to the initial biosimilars used in supportive care, trastuzumab is an anticancer drug, a specific targeted agent and thus it appears that the neoadjuvant setting may be the right choice to compare a trastuzumab biosimilar with the originator to assess efficacy and safety [Jackisch et al. 2015]. Several trastuzumab biosimilars are either ready to be commercialized or are in clinical development [Hurst et al. 2014]. Two trastuzumab biosimilars are discussed below, PF-05280014 (Pfizer®) and CT-P6 (Celltrion®).
PF-05280014
The agent PF-05280014 is a potential trastuzumab biosimilar. The agent was compared with trastuzumab [Herceptin® marketed in the US (trastuzumab-US) and European Union (trastuzumab-EU] [Hurst et al. 2014]. Preclinical studies were designed to compare the similarity of PF-05280014 to the originators. In vitro structural and functional analysis, and in vivo PK and immunogenicity were assessed for comparability. Preclinical in vitro studies comprised peptide mapping and tumour cell growth inhibition assays. In vivo studies were performed using mice receiving a single dose (0, 1, 10 or 100 mg/kg) of PF-05280014, trastuzumab-US or trastuzumab-EU. The mice were monitored over a 4 month period. PF-05280014, trastuzumab-US and trastuzumab-EU were well tolerated throughout this time. PF-05280014, trastuzumab-US and trastuzumab-EU demonstrated similar structures, similar tumour cell growth inhibition properties and similar PK profiles. The incidence of ADA was very low and similar across the three MoAbs. The authors concluded that their results support the clinical development of PF-05280014 as a Herceptin® biosimilar [Im et al. 2013].
Recently, the same trastuzumab biosimilar, PF-05280014, was evaluated in a phase I, double-blind trial [ClinicalTrials.gov identifier: NCT01603264] [Yin et al. 2014]. This study was performed on 105 healthy male volunteers. The subjects were randomized 1:1:1 to receive a single 6 mg/kg, IV dose of PF-05280014, trastuzumab-EU or trastuzumab-US, and evaluated for 70 days. Drug concentration–time data were analysed by noncompartmental methods. The PK similarity of the biosimilar, PF-05280014, to each of the trastuzumab originators was determined using the standard 80.00–125.00% bioequivalence criterion. A total of 101 subjects were studied and the PK evaluated. The three agents (PF-05280014, trastuzumab-EU, and trastuzumab-US) exhibited similar PK profiles with target-mediated disposition. Adverse events (AEs) were similar across all arms with TEAEs reported by 71.4%, 68.6% and 65.7% subjects in the PF-05280014, trastuzumab-EU and trastuzumab-US arms, respectively. The most common AEs were infusion-related reactions, headache, pyrexia, nausea and chills. All post dose samples except one tested negative for ADA. The authors of the study concluded that the three agents, PF-05280014, trastuzumab-EU and trastuzumab-US, had similar PK as well as comparable immunogenicity and safety profiles [Yin et al. 2014].
CT-P6
CT-P6 is new trastuzumab biosimilar currently undergoing clinical development. The results of a phase III, double-blind, randomized, parallel group trial to demonstrate the equivalence in efficacy and safety of CT-P6/paclitaxel and Herceptin®/paclitaxel in metastatic breast cancer (MBC) patients were recently reported [EMA, 2012]. The patients included in the study were HER2+ by fluorescent in situ hybridization (FISH) with measurable disease, having received chemotherapy for MBC for at least 12 months and no prior trastuzumab treatment. The patients were treated with adjuvant/neoadjuvant trastuzumab and chemotherapy. The total number of patients included was 475: 244 in the paclitaxel + trastuzumab + CT-P6 arm, every 3 weeks, and 231 patients in the paclitaxel + trastuzumab arm, also every 3 weeks. Eastern Cooperative Oncology Group (ECOG) status at entry into the protocol was 0–1. Exclusion criteria were: prior chemotherapy for MBC; central nervous system (CNS) metastasis; and a baseline left ventricular ejection fraction (LVEF) ⩽50% or history of chronic heart failure (CHF).
The results of the study demonstrated a CR of 3.7% versus 1.7% for CT-P6/paclitaxel and trastuzumab/paclitaxel by an independent committee (ITRC), and 4.9% versus 3.6% by the researcher. In terms of partial response (PR), the results were 52.9% and 60.2% by the ITRC, and 59.2% and 65.8% by the researcher, respectively. In terms of stabilization of the disease (SD), the results were 20.1% and 16.5% by the ITRC, and 25% and 24.2% by the researcher, respectively. Finally, the overall response rate (ORR) results were 56.6% and 61.9% by the ITCR, and 64.8% and 58.4%, by the researcher, respectively [Im et al. 2013]. Statistical analysis [full analysis set (FAS) and per patient set (PPS)], using the difference in the proportion of CR or PR with a confidence interval estimated using the exact method, all favoured the trastuzumab/paclitaxel arm. The time to progression at 1 year did not show any statistical difference (p = 0.0978). CT-P6 was well tolerated and had an excellent safety profile, similar to that of trastuzumab, the originator [EMA, 2012].
The aim of clinical trials with trastuzumab biosimilars is to show equivalence, and not patient benefit, as this was shown with Herceptin®. Once biosimilars of trastuzumab are approved, several challenging issues will need to be addressed, such as maintaining appropriate pharmacovigilance, extrapolating across indications, and automatic substitution and switching. No consensus has yet been reached in any of these areas.
Discussion
Biosimilars, new agents manufactured after the patent covering the originator has expired, are here to stay. This is clear from the positioning of the regulatory agencies, both in Europe and the United States. Oncologists want the best products on the marked in terms of efficacy and safety. So far, at least from evaluating the past five years of biosimilars on the European market, they appear to fulfil those expectations. The difference now is in the nature of the agents. Early biosimilars were mostly used in supportive care, with fast, immediate responses, making the evaluation of parameters, efficacy and safety relatively straightforward. Now the situation has changed dramatically, since the new wave of biosimilars are intended to treat cancer and the issue of patient outcome becomes a priority; in particular, when responses can be so heterogeneous across oncology populations.
It appears clear that future studies of these new targeted compounds should be performed in a sensitive and homogeneous population. Biosimilars should be studied in the particular population of patients for whom, if there is a difference between the biosimilar and reference product, that difference will most easily be detected and we must be selective. At least for breast cancer, the neoadjuvant/adjuvant patient population must be, for example, a homogeneous and sensitive population to establish the similarity of Herceptin® and the biosimilar trastuzumab. Nevertheless, the neoadjuvant would be the preferred option to assess both efficacy and safety, since it can provide answers much faster. The adjuvant setting would take years to detect a clinical difference in the response between the biosimilar and the originator. However, the field faces many other challenges; for instance, how to distinguish loss of clinical response due to natural progression (unavoidable) from neutralization by anti-MoAb antibody response (potentially treatable).
The EMA guidelines for MoAbs in clinical trials [EMA, 2012] identify the ORR as a sufficiently sensitive endpoint for clinical trials of biosimilar antibodies. However, very often the ORR does not correlate to survival. Common endpoints in clinical trials, such as OS or PFS, may provide superior data but are not necessarily convenient for a biosimilar antibody trial [Bui and Taylor, 2014].
The results from the phase III clinical trial using the trastuzumab biosimilar CT-P6 [Im et al. 2013] has provoked some concern within the oncology community. Although the sample size is small, 244 versus 231, still in terms of efficacy the trastuzumab biosimilar came up short compared with the originator. This is probably unfair to the biosimilar, but the debate is now open. Do we accept a range of variability of 10%, or 15% as was accepted for generics? If so, and since we are now dealing with an anticancer effect, is this acceptable? Some researchers say that, with a larger sample, these small differences would probably have disappeared. We know from studies that go back 30 years that even a reduction in the dose of 15% can have important effects on the final outcome for the patient. We are no longer dealing, as in the past, with supportive care biosimilars but with anticancer compounds, which is a completely different story. The debate will probably continue until more data are collected that can reassure clinicians one way or another.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The author declares no conflicts of interest in preparing this article.