
Abstract
Background
Influenza is a disease of significant morbidity and mortality, the number of anti-influenza drugs is small; many of them stimulate the appearance of resistant strains. In this work, we demonstrate activity of some usnic acid (UA) derivatives against influenza virus in vitro and in vivo.
Methods
Organic synthesis was used to prepare compounds. Antiviral activity of the compounds in vitro was evaluated by their ability to decrease the virus titer on Madin–Darby Canine Kidney cells. In vivo activity was evaluated by decrease of mortality and index of protection.
Results
Compounds were tested against a broad spectrum of influenza virus strains and showed activity against all used strains. One compound,
Conclusion
Our results suggest that valine enamine of UA could be a potential candidate for the development of a new anti-influenza therapy.
Introduction
Influenza A virus is a human pathogen of primary importance for health-care systems all over the world and represents serious problem for drug development, vaccination, and treatment strategies. 1
Three chemical classes of compounds against different viral targets are currently used as anti-influenza drugs. First, they include adamantane derivatives represented by two compounds: rimantadine (α-methyl-1-adamantylamine hydrochloride) and amantadine (1-aminoadamantane). These compounds block the virally encoded protein M2, which acts as a proton channel required for acidification of the virion core. This disrupts the M–RNP interaction and enables the core to dissociate after membrane fusion has released it into the cytoplasm. 2 Second, there are four neuraminidase inhibitors: zanamivir (Relenza®), oseltamivir (Tamiflu®), peramivir (Rapiacta®), and laninamivir (Inavir®). 3 They interfere with the viral neuraminidase, which plays an essential role in the release of progeny virus particles from the surface of a host cell. Third, nucleoside analogs ribavirin and favipiravir are antivirals of broad range of activity, which exhibit a suppressive effect against almost all RNA-genome human viruses. 4 However, ribavirin, a nucleoside analog, possesses numerous side effects, including the reduction of hemoglobin level, neutropenia, pulmonary edema, and so on. 5
Influenza virus is a dynamic pathogen with rapid selection of drug-resistant strains. Indeed, adamantane resistance among circulating influenza A viruses increased rapidly worldwide in 2003–2004. 6 In this regard, one more example should be mentioned where pre-pandemic influenza viruses of H1N1 subtype resistant to oseltamivir, effective, and internationally accepted anti-influenza drug, emerged and spread worldwide since 2007–2009 having resulted in total resistance (100% of tested strains). 7 These facts underscore the serious challenge for search and development of novel anti-influenza drugs with broad range and alternative mechanism of activity.
Previously, we showed anti-viral properties of usnic acid (UA) and its derivatives against influenza virus A/California/07/09 (H1N1)pdm09.8,9 In the present work, we describe the spectrum of anti-influenza activity of the most effective compounds in this group, provide evidence for their protective activity in animals, and hypothesize about their mode of action.
Materials and methods
Compounds
Viruses and cells
Madin–Darby Canine Kidney (MDCK) (ATCC # CCL-34) cells were maintained in alpha minimum essential medium (MEM) supplemented with 5% fetal bovine serum (FBS) and ciprofloxacin (2 µg/ml). Influenza viruses A/Puerto Rico/8/34 (H1N1), A/California/07/09 (H1N1)pdm09, A/Vladivostok/2/09 (H1N1), and A/Aichi/2/68 (H3N2) were obtained from the collection of viruses of the Influenza Research Institute. Prior to an experiment, viruses were propagated in the allantoic cavity of 10- to 12-day-old chicken embryos for 48 h at 36℃ (influenza A viruses) or 72 h at 34℃ (influenza B virus). Infectious titer of the virus was determined in MDCK cells in 96-well plates.
Animals
Inbred female mice, 12–16 g were obtained from the animal breeding facility of Russian Academy of Medicine “Rappopolovo” (Rappopolovo, Russia). The mice were quarantined one week prior to the experimental manipulation and were fed standard rodent chow with ad libitum access to water. Animal experiments were conducted in accordance with the principles of laboratory animals care 11 and were approved by the Institutional Ethical Committee.
Cell viability assay
For the compound’s cytotoxity evaluation, MDCK cells were seeded in 96-well plates and cultivated in Eagle’s MEM with addition of 5% fetal calf serum. The compounds were dissolved in dimethyl sulfoxide (DMSO) to 5 μg/ml, and serial twofold dilutions (1000–4 µg/ml) were prepared in MEM. After the monolayer formation, the cells were washed by serum-free MEM, the dilutions were applied to the cells and incubated for 48 h at 37℃. After incubation, the cells were washed twice with phosphate-buffered saline (PBS) and the number of surviving cells were evaluated by a microtetrazolium test (MTT). Briefly, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (ICN Biochemicals Inc., USA, 0.5 μg/ml, 0.1 ml per well) was added with subsequent incubation of plates at 37°С in 5% CO2 for 60 min. The colored precipitate deposit was dissolved in 100 ml of DMSO. The plates were swirled gently and left in the dark at room temperature for 30 min. Optical density was measured using a spectrophotometer (Victor 1420, Perkin Elmer, Finland) at wavelength 535 nm. Based on this data, the value of compound concentration required to reduce 50% cell viability (50% cytotoxic concentration, hereafter referred to as CTD50,) was calculated for each compound.
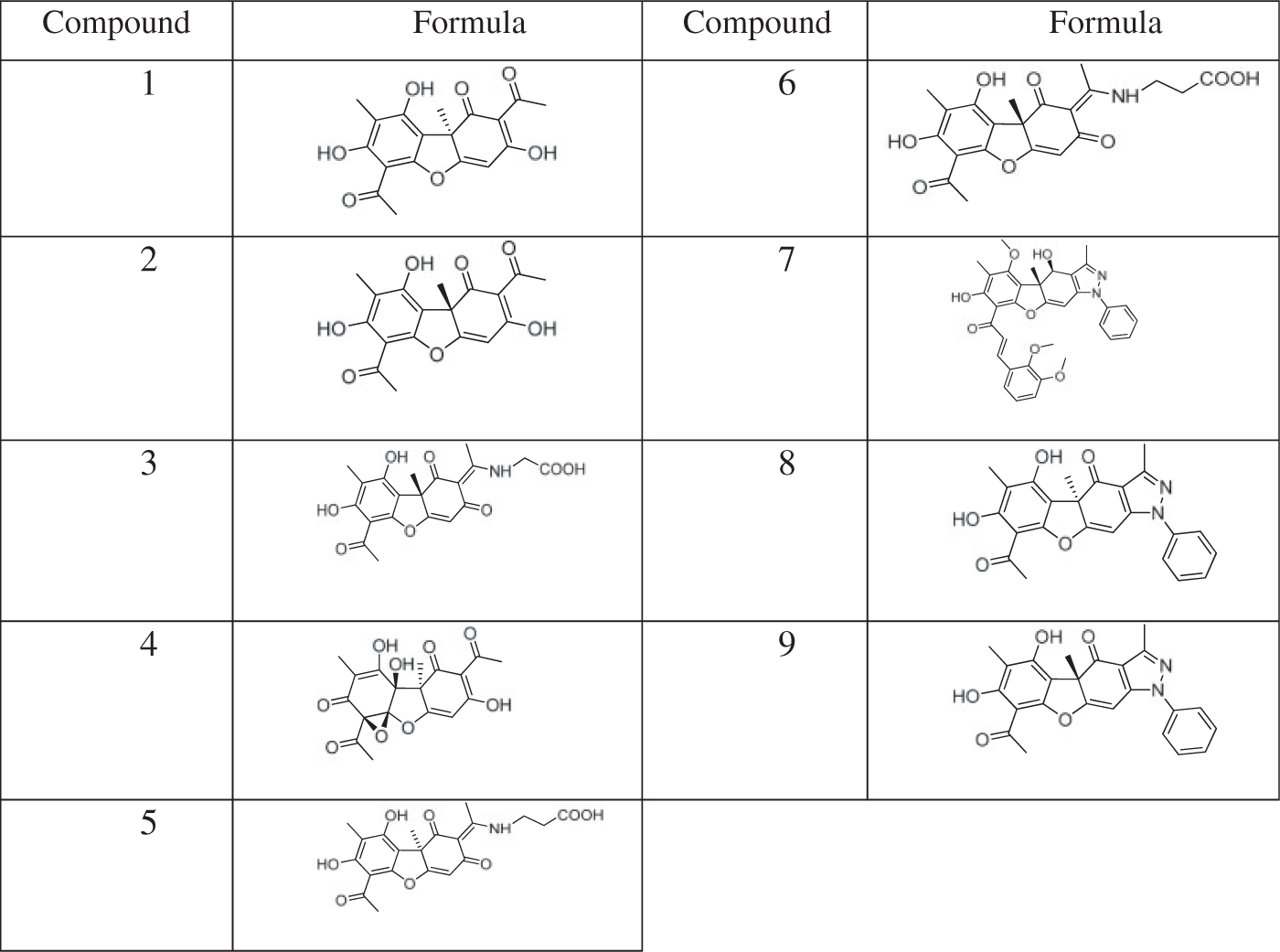
Inhibitory effect of UA derivatives against broad-ranging influenza viruses
Serial twofold dilutions of compounds were prepared in MEM (Biolot, Russian Federation) containing 2 mM arginine (Sigma–Aldrich, USA), 2 mM glutamine (Sigma–Aldrich, USA), and 2 µg/ml trypsin (Sigma–Aldrich, USA), and incubated with MDCK cells for 1 h at 36℃ in presence of 5% CO2 (volume 100 µl per well). Each concentration was tested in triplicate; untreated cells were used as control. The cell culture was then washed twice with PBS and inoculated with appropriate virus at multiplicity of infection 0.01 (volume 100 µl per well) and appropriate compound dilution (volume 100 µl per well; total volume of each well was 200 µl) and incubated for 24 h. After incubation, tenfold dilutions of the supernatant were added to MDCK cells and then incubated for 48 h at 36℃ in presence of 5% CO2. A virus titer in the supernatant was determined by hemagglutination assay.
Hemagglutination assay was carried out as follows. One hundred microliters of supernatant was transferred into round bottom wells and mixed with 100 µl of 1% suspension of chicken erythrocytes and then incubated for 1 h at room temperature. The virus titer was considered as a reciprocal to the final dilution of the inoculum in which the virus was able to cause positive hemagglutination in 50% of wells and expressed in 50% tissue culture infecting doses (TCID50). Antiviral activity of the compounds was evaluated by their ability to decrease the virus titer. Based on the results, the IC50, 50% inhibiting dose (concentration of compound that decreases the virus production twofold comparing to control), was calculated. Selectivity index (SI) was calculated as relation of CTD50 to IC50. All the compounds having SI ≥ 10 were declared active.
Virus lethality titration
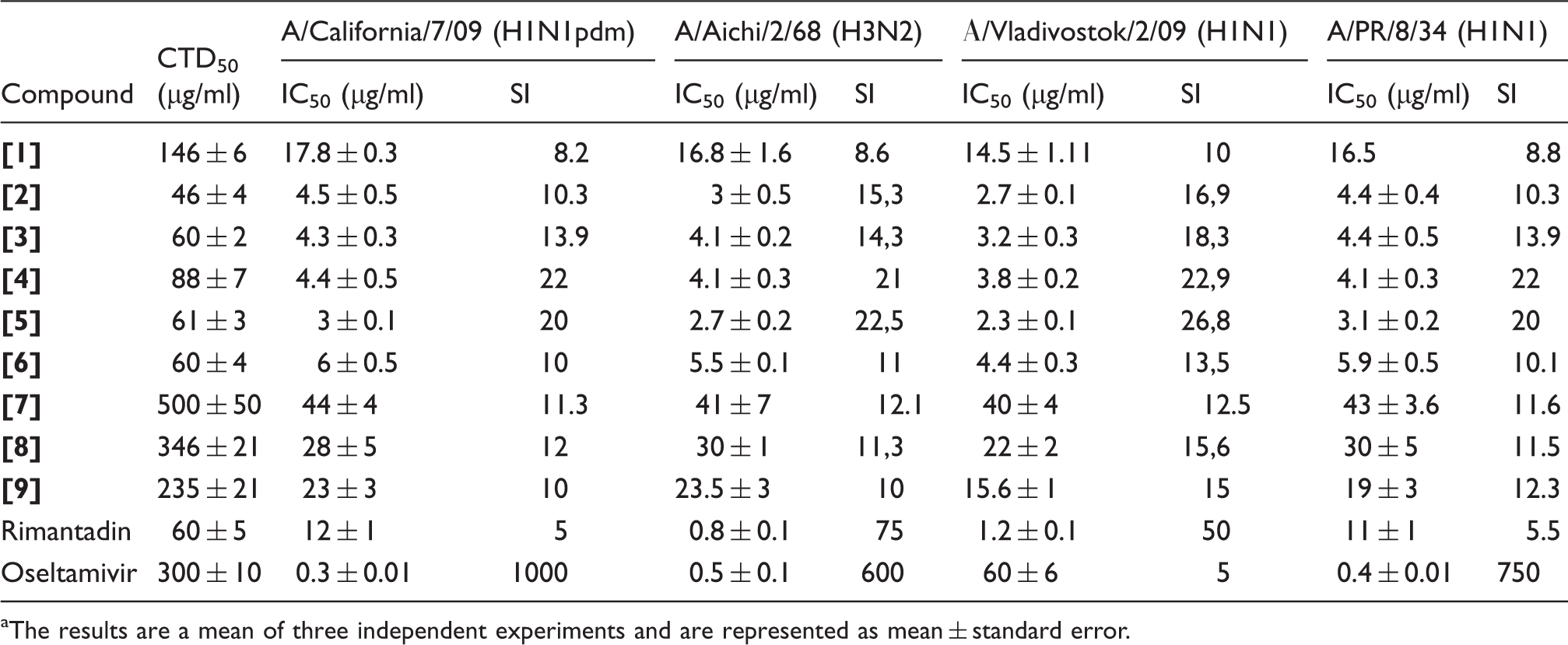
Prior to studies of the protective activity of compounds in animals, mouse-adapted influenza virus was titrated for lethal effect. For this purpose, mice (10 in each experimental group) were inoculated intranasally under anesthesia with 50 µl of serial decimal dilutions (10−1 to 10−5) of the lung homogenate from virus-infected mice. The dilution that caused death of 50% of the animals in 21 days postinfection (LD50) was calculated as described previously 12 and was used for subsequent experiments.
In vivo experiments
In order to evaluate anti-influenza activity of compounds in vivo, mice (15 per group) were infected with one LD50 of previously titrated virus (see Virus lethality titration section). Compounds were previously studied for determine their 50% lethal toxic doses (LTD50) and then administrated intraperitoneally in a dose 1/5 LTD50 once a day in a volume of 0.2 ml for days 1–4 postinfection. Control animals were treated with saline solution. Ten uninfected untreated mice were used as intact control.
In each group, 10 mice were used for mortality control. Each group was monitored daily on lethal cases for two weeks postinoculation. Based on the data received, percentage of mortality and index of protection ((LE − LC)/LE)*100% (LE: lethality in the experimental group; LC: lethality in the control group) were calculated.
Histological examination
Lungs of mice were placed into 10% PBS-buffered formaldehyde, dehydrated in graded ethanol, and embedded in paraffin. Four-micrometer slices were prepared and stained with hematoxylin–eosin.
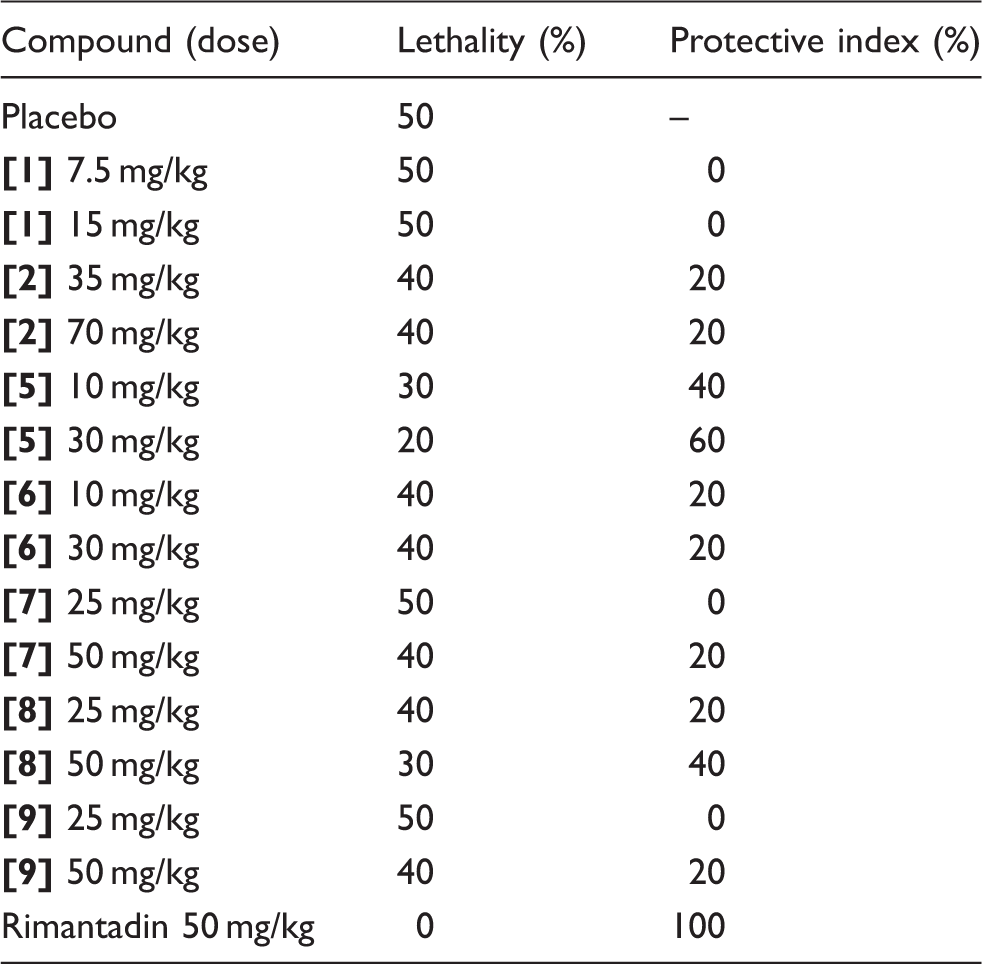
Cultivation of resistant strains
Influenza strain A/PR/8/34 (H1N1) was cultivated in MDCK cells in presence of growing concentrations of compounds, beginning with one IC50 to half CTD50 during 13 passages. The criteria for identifying resistance depended on comparing the antiviral effect of the compound against the original strain and the strain after repeated passage in the presence of the compound.
Molecular biology methods
RNA was isolated using QIAGEN Rneasy TotalTM RNA Isolation Kit according to the manufacturer s instructions. Reverse transcription was carried out using SuperScript III One-Step RT-PCR System (Invitrogen, USA) for 45 min. Further amplifications were carried out with a Thermocycler C1000 using the following amplification program: 94℃—2 min, 50℃—30 s, 72℃—1 min (30 cycles).
Amplification products were analyzed by gel-electrophoresis in 2% agarose gel. DNA was then extracted using QiaQuick Gel Extraction Kit (Qiagen, Germany). Sanger sequencing was carried out using ABI BigDye Terminator v. 3.1 Cycle Sequencing Kit (Applied Biosystems). Fragment assembly was performed with the program pack Vector NTI 10 Advance (Invitrogen, USA).
Statistical analysis
Results are represented as means ± standard errors of the mean. The values of CTD50 and IC50’s were calculated using PRISM 6.0 software. Animal survival was analyzed by log-rank (Mantel–Cox) test.
Results
Antiviral activity of UA derivatives in vitro
The spectrum of antiviral activity of usnic acid derivatives. a
The results are a mean of three independent experiments and are represented as mean ± standard error.
As evidenced, the activities of compounds against different viruses were similar, in general. It is also evident from the presented data that the activities of all tested derivatives of UA were lower than the reference substances, except resistant strains.
In vivo experiments
Next, the activities of usnic acid derivatives were studied in vivo. For the experiment compounds
At first, LTD50 (50% toxic dose) was identified in preliminary experiments. For compounds
Protective activity of usnic acid derivatives in vivo against influenza virus A/Aichi/2/68 (H3N2), viral dose is one LD50.
Data presented in Table 2 indicate that all examined compounds reduce mortality and increased the mean lifetime of the animals comparatively to the placebo group. The most statistically significant difference (p < 0.05) was shown for the compound
Selection and study of resistant strains
Antiviral activity of compound [5] against final and original strains of A/PR/8/34 (H1N1).
As can be seen from the table, viral titer decrease was the same at the same doses. Thus, after 13 passages with compound
In spite of absence of resistant strains, the possibility of any mutations genesis still remained, so we sequenced genes NA, HA, and M2, after what sequences were compared. No genes had any substitutions comparatively to wild type.
Study of hepatotoxicity of compound [5]
Past evidence of liver toxicity for UA prompted the examination of the livers from mice treated with
In the liver of untreated animals (Figure 1a), lobules of liver were presented with central veins with radiating hepatic tubules. Hepatocytes remained intact; the cytoplasm was homogeneous or weakly vacuolated. No signs of cell degradation or inflammatory infiltration were seen.
(a) Hepatic lobule of untreated mouse. Hepatic tubules are normal, cell degradation is absent. (b) Liver tissue of mouse treated by usnic acid (30 mg/kg). Severe degradation of hepatocytes, intensive infiltration of neutrophils. (c) Hepatic lobule of mouse treated by compound 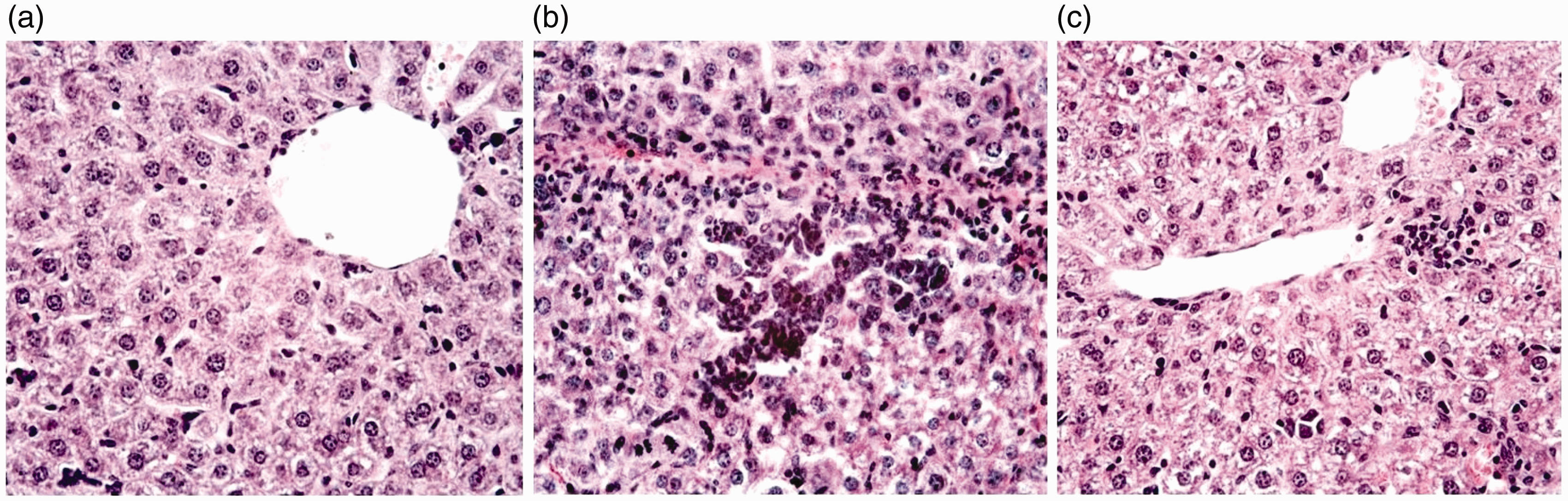
Hepatic parenchyma of animals treated with UA (Figure 1b) exhibited hepatocytes at various stages of degradation, including cytoplasmic vacuolation. Foci of cell disruption and polymorphonuclear leukocyte infiltration were found frequently. These observations are typical of the toxic liver disease and indicate a high hepatotoxicity of UA.
Signs of hepatotoxicity in mice treated by compound
Based on results obtained, we can make a conclusion that valine modification of UA can reduce its toxicity for liver tissues and, at the same time, increase its antiviral properties. Nevertheless further investigations of reducing hepatotoxicity of UA derivatives are required.
Discussion
In spite of the existence of antiviral drugs and vaccines, influenza is a cause of significant morbidity and mortality, posing a serious health threat during seasonal outbreaks and periodic pandemics. Furthermore, influenza viruses are capable of developing drug resistance very quickly, making effective antivirals useless. Use of plant metabolites as parent compounds for creating of novel biologically active agents is an established practice in modern medical chemistry.
UA is produced as a secondary metabolite by lichens of genus Usnea. Due to high accessibility and easy purification, UA was previously tested for various types of biological activity. In particular, its wound healing, 13 antimicrobial, 14 antimycobacterial,15,16 antiprotozoal, apoptosis-inducing and antiproliferative, 17 anti-inflammatory, and analgesic 18 activity was demonstrated. Its antiviral action was also shown against type 1 herpesvirus, 19 human papillomavirus, 20 Epstein–Barr virus, 21 polyomavirus, 22 and arenaviruses. 23
Little data are available about the biological activity of UA derivatives. Among them, enamine derivatives were studied regarding their cicatrizing properties.
24
The compound
As far as we know, no information is available about antiviral properties of UA derivatives. Previously,8,9 we studied anti-influenza activity of 88 compounds of this group. The most effective of them confirmed their efficacy in animal model of influenza infection conducted in this study.
In the present work, we showed a wide range of activity of UA derivatives against influenza viruses differing in their antigenic type and subtype, susceptibility to current antivirals and host specificity. We demonstrated that compounds of this group suppress replication of a broad panel of influenza viruses.
Derivatives of UA showed moderate activity in experiments in vivo. The most active compound was
In order to examine some details about the mechanism of action of the derivatives of UA selection of viral strains resistant to the compound
Because of well-known hepatotoxicity of UA, histological examination of liver tissue of mice treated with UA and compound
In summary, we can say that UA derivatives have a broad spectrum of anti-influenza activity in vitro and one of them, compound [5], also showed moderate activity in vivo. This compound does not stimulate the selection of resistant strains and its hepatotoxicity was reduced comparatively to UA. Our results suggest that valine enamine of UA could be a potential candidate for the development of a new anti-influenza therapy.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.