
Abstract
Rare tumors are a heterogeneous group of neoplasms with low incidence and are usually difficult to diagnose. Certain tumors that are common at specific anatomical locations rarely occur at unexpected locations and cause diagnostic challenges. Although histopathology is central to the diagnosis of these lesions, as with any tumor, ancillary techniques such as immunohistochemistry and molecular studies are often essential. In addition, a multidisciplinary approach that includes clinical, radiological, and biochemical inputs plays an important role in achieving an accurate diagnosis. A total number of 10 benign and malignant neoplastic lesions at rare locations were analyzed. Immunohistochemistry was performed for the required cases. The 10 cases included were: primary nasal meningioma, malignant melanoma of the lacrimal sac, metastatic synovial sarcoma of the thyroid, malignant teratoma of the thyroid, papillary serous cystadenocarcinoma of the paratestis, leiomyoma of the testis, squamous cell carcinoma arising in an epidermoid cyst, intraarticular synovial sarcoma of the knee, Ewing sarcoma of the kidney, and primary plasmacytoma of the breast. Knowledge and awareness of these entities improve the quality of reporting in any individual case. As a group, these lesions require further studies to enhance the understanding of their pathogenesis and patient care.
Keywords
Introduction
Neoplasms represent a heterogeneous group of lesions that arise from aberrant gene expression. These tumors originate from a single mutated cell, probably from a stem cell, proliferate and differentiate in a manner that reflects the native tissue from which they arise. The diagnosis of these tumors is straightforward in typical locations when they exhibit distinctive morphological features. However, identifying similar tumors in unusual locations presents challenges for both clinicians and pathologists. Rare tumors or diseases are those, that occur in a small proportion of patients, and rare cancers are defined as having an incidence of fewer than six cases/100,000/year.1,2 As a group, these account for 22% of all cancers, which is higher than that of any single common cancer, causing a significant burden on the healthcare system.1,2 This group includes not only tumors of low incidence, but also common tumors occurring in uncommon locations, which equally pose diagnostic challenges. Moreover, unlike rare tumors, commonly occurring tumors at uncommon sites have not received significant attention. The principal challenge in addressing such tumors is the possibility of misinterpretation due to lack of expertise. Limited clinical trials and research on these tumors have resulted in restricted therapeutic options and poor patient outcomes. Accurate diagnosis of these tumors often necessitates ancillary techniques, such as immunohistochemistry and molecular genetics. 3 Recent advances in genome sequencing, RNA sequencing, and omics analysis in combination with conventional methods have enabled an integrated approach for the study of these rare tumors. Such integration has the potential to enhance treatment options and improve patient outcomes. 3
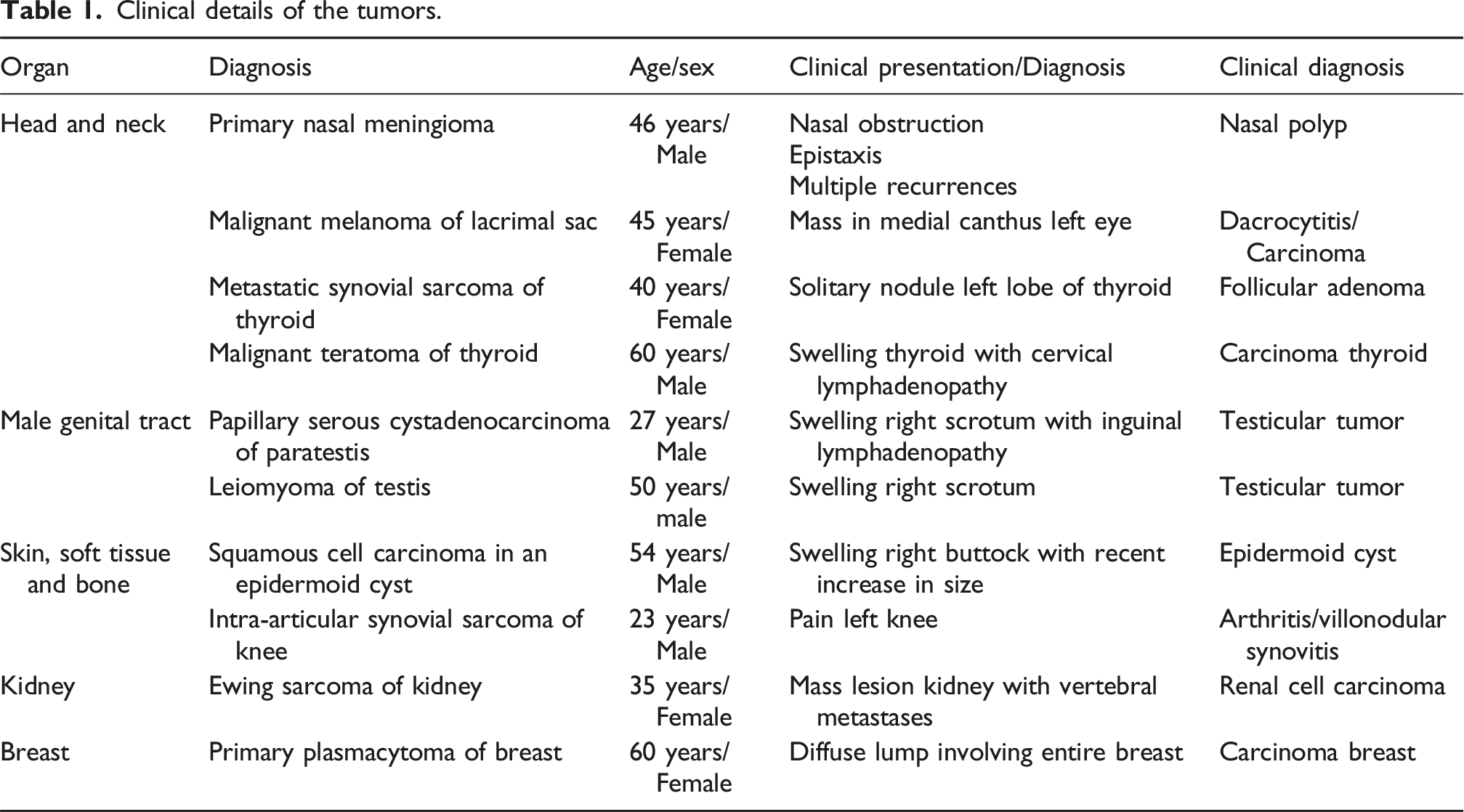
Clinical details of the tumors.
Case details
Head and neck tumors
Case 1: Primary nasal meningioma
A 46-year-old male presented with recurrent episodes of nasal obstruction and epistaxis for 5 months. Computed tomography (CT) scan of the paranasal sinuses showed a well-defined hypodense lesion in the right nasal cavity with no intracranial connection. Rhinoscopy revealed a well-defined mass lesion at the base of septum in the right nasal cavity. Histopathology showed an unencapsulated lesion composed of sheets of nondescript oval shaped cells exhibiting moderate pleomorphism, occasional mitotic activity and focal mucin. The overlying respiratory epithelium was intact. The undifferentiated nature of the neoplastic cells and focal mucin prompted us to initially diagnose the patient with poorly differentiated carcinoma. However, the focal whorling pattern and occasional intranuclear inclusions made us to consider the diagnosis of meningioma. Immunohistochemistry revealed that the tumor cells were negative for CK, and positive for vimentin and EMA, confirming the diagnosis as meningioma (Figure 1). Photomicrograph of primary nasal meningioma. (a). Sheets of neoplastic cells beneath intact respiratory epithelium, H & E stain, 200x. (b). Sheets of meningothelial cells with occasional intranuclear inclusions, H & E stain, 400x. (c). Tumor cells showing strong cytoplasmic positivity for vimentin, 400x. (d). Tumor cells showing cytoplasmic positivity for epithelial membrane antigen (EMA), 400x.
Case 2: Malignant melanoma of lacrimal sac
A 45-year-old female patient presented with a mass lesion located in the medial canthus of the left eye, associated with intermittent epiphora for a period of 1 month. The patient also reported a single episode of bloody discharge from the eye. An excision biopsy of the tumor revealed two dark brown solid and cystic masses containing dark brown fluid. Microscopy showed a circumscribed nodular lesion in continuity with the lacrimal sac epithelium. The lesion was composed of fascicles of spindle cells and sheets of polygonal cells, both with vesicular nuclei, prominent nucleoli, and intracytoplasmic brown pigment (Figure 2). A diagnosis of malignant melanoma was established. Immunohistochemistry and confirmatory molecular testing was denied by the patient. Photomicrograph of malignant melanoma of the lacrimal sac. (a). Subepithelial lesion composed of sheets of pleomorphic cells with intracytoplasmic pigment, H&E stain, 100x. (b). Pleomorphic spindle-shaped cells, H&E stain,100x. (c). Tumor cells with melanin pigment, H&E stain, 200x and (d). Pleomorphic tumor cells with melanin pigment. H&E,400x.
Case 3: Metastatic synovial sarcoma of thyroid
A 40-year-old female presented with a swelling in front of the neck for about 2 months. A 3 × 2 cm nodule involving the left lobe of the thyroid was observed on clinical examination. Fine needle aspiration cytology (FNAC) was performed, and the cellular cytology smears showed oval and spindle shaped cells in clusters and discretely. A cytological differential diagnosis of medullary carcinoma/mesenchymal tumor was considered. A 7 × 4 × 3 cm total thyroidectomy specimen received, which on sectioning showed a 3.5 × 2 × 1 cm well-defined gray-white mass in the left lobe. Histopathology revealed a thick-capsulated cellular tumor of pleomorphic oval and spindle cells with high mitotic activity. The tumor showed focal infiltration into the adjacent thyroid tissue. The two differential diagnoses considered were medullary carcinoma and spindle cell sarcoma. On immunohistochemistry (IHC), the tumor cells were immunoreactive to EMA, calponin, BCL2, and TLE1 and negative for calcitonin, CEA, SMA, desmin, S100, CD34, TTF1, and thyroglobulin. Upon further questioning, the patient recalled a previous surgical procedure for a tumor in the thigh region. Based on these findings, a diagnosis of metastatic synovial sarcoma was established (Figure 3). Molecular study was not planned due to financial constraints. Photomicrograph of metastatic synovial sarcoma of the thyroid. (a). Encapsulated cellular lesion with normal thyroid at the periphery, H & E stain, 100x. (b). Cellular lesion composed of fascicles of spindle-shaped cells, H & E stain, 200x. (c). Sheets of pleomorphic oval-shaped cells, H&E stain, 400x. (d). Strong nuclear positivity for transducin-like enhancer of split 1 (TLE1), 400x.
Case 4: Malignant teratoma of thyroid
A 60-year-old male presented with a swelling in front of the neck for 2 years with recent rapid growth and pressure symptoms. Ultrasound and CT scans of the neck identified a solid and cystic mass involving the thyroid gland, associated with cervical lymphadenopathy. Suspecting thyroid cancer, total thyroidectomy with modified radical neck dissection was carried out. The gross specimen measured 10 × 6 × 4 cm, and the cut section showed a variegated tumor with solid and cystic components involving the entire thyroid. Microscopic examination of the thyroid and cervical lymph nodes showed features of a malignant teratoma, composed of immature tissue derived from all three germ layers, including primitive neuroectodermal tissue. (Figure 4). Photomicrograph of primary teratoma of the thyroid. (a). Primitive neural tubules, H & E stain, 200x. (b). Residual thyroid follicles surrounded by tumor, H & E stain, 100x. (c). Immature cartilage, H & E stain, 200x. (d). Adjacent normal thyroid tissue, H & E stain, 100x.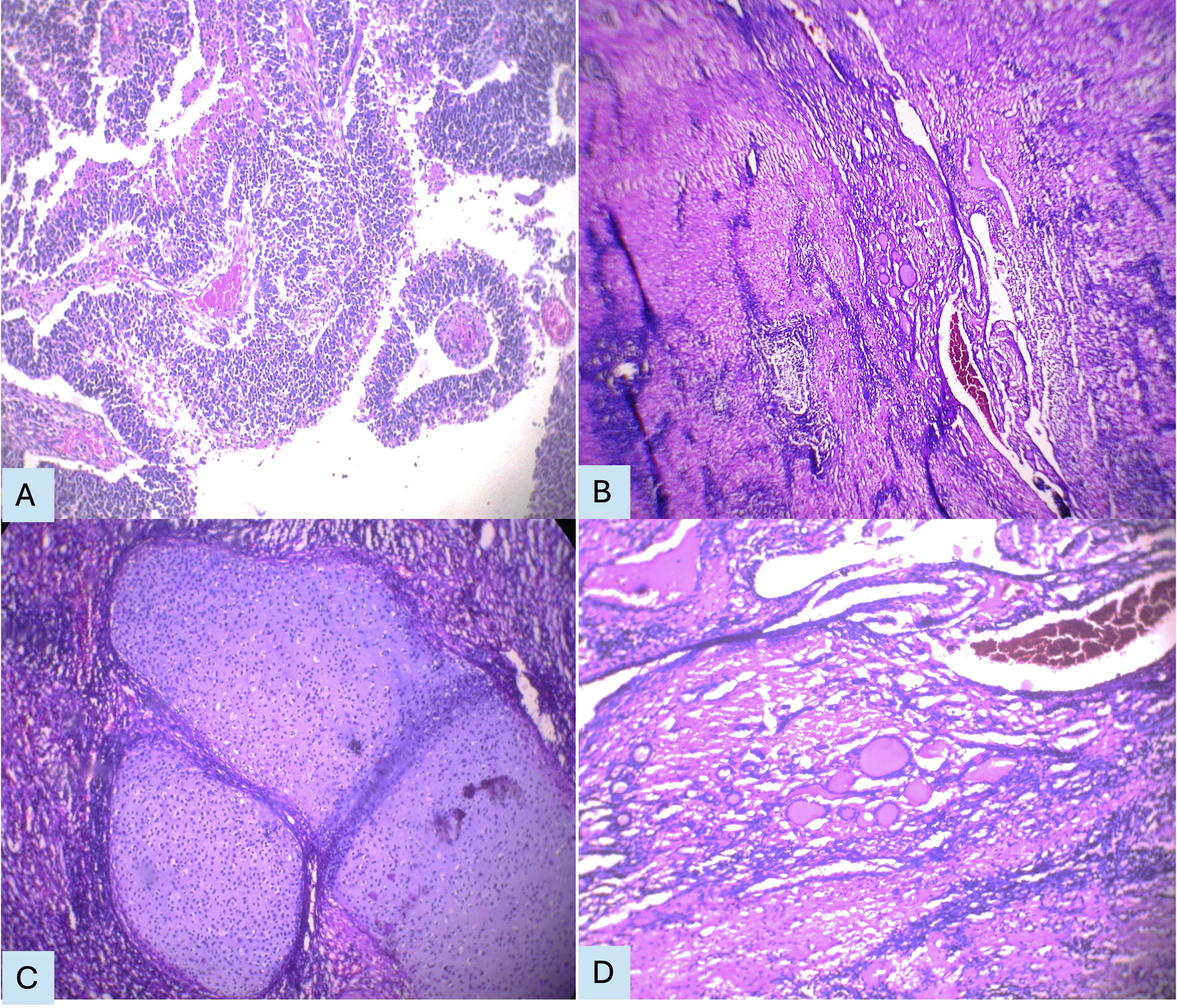
Male genital tract tumors
Case 5: Papillary serous cystadenocarcinoma of paratestis
A 27-year-old male presented with swelling of the right half of the scrotum and bilateral inguinal lymphadenopathy for 6 months. Ultrasonography of the scrotum was reported as an inflammatory collection. FNAC of the inguinal lymph node suggested papillary carcinomatous deposits. A right high orchidectomy specimen revealed a 6 × 4 × 2 cm gray-white friable growth beneath the thickened sac, with the testis remaining uninvolved. Microscopy showed a paratesticular papillary tumor with branching fibrovascular cores and numerous psammoma bodies, infiltrating the testis and epididymis at few foci. The two differential diagnoses considered were malignant mesothelioma and papillary serous cystadenocarcinoma. Of the immune markers used, calretinin, WT1, and PAX8 were negative, while CEA was focally positive and CK 7 exhibited strong membrane positivity (Figure 5). The patient was diagnosed with papillary serous cystadenocarcinoma of the paratestis. Photomicrograph of papillary serous cystadenocarcinoma of paratestis. (a). Normal testis with a paratesticular papillary tumor, H & E stain, 200x. (b). Papillae with fibrovascular cores, multilayered epithelial cells, and psammoma bodies, H & E stain, 400x. (c). Strong membrane positivity for cytokeratin 7 (CK7), 400x. (d). Focal positivity for carcinoembryonic antigen (CEA), 400x. (e). Negative staining for Wilms tumor 1 (WT1), 200x. (f). Negative staining for calretinin, 200x.
Case 6: Leiomyoma of paratestis
A 50-year-old male presented with a painful swelling of the right half of the scrotum for 1 month. A high orchidectomy was done with a clinical suspicion of malignancy. The gross specimen measured 4 × 3 × 2 cm, and the cut section showed a nodular gray-white paratesticular mass compressing the uninvolved testis. Microscopy demonstrated a circumscribed tumor with short and long fascicles of bland spindle cells with eosinophilic cytoplasm. The tumor was focally continuous with the tunica albuginea, and thick walled blood vessels were found adjacent to it. The testis and epididymis were uninvolved. Immunohistochemistry with calponin was positive, confirming the diagnosis of leiomyoma.
Skin and soft tissue tumors
Case 7: Squamous cell carcinoma in an epidermoid cyst
A 54-year-old male reported to the surgical out-patient department with a swelling on his right buttock for 3 years with a recent rapid increase in size. The tumor was a 8 × 6 cm firm, fixed, and nodular mass with intact skin. Excision biopsy showed a 10 × 7 × 4 cm partly skin covered mass. On sectioning, a five cm diameter cyst filled with pultaceous material surrounded by gray-white infiltrative growth was found. Microscopy disclosed the typical features of an epidermoid cyst with a squamous lining cavity containing pultaceous material. It was surrounded by an infiltrative squamous cell carcinoma in continuous with squamous cell carcinoma in situ of the lining epithelium of the cyst (Figure 6). A diagnosis of squamous cell carcinoma arising from an epidermoid cyst was established. Photomicrograph of squamous cell carcinoma arising in an epidermoid cyst. (a). Cyst filled with keratin surrounded by squamous cell carcinoma, H & E stain, 100x. (b). Infiltrating tumor arising from a keratin-filled cyst, H & E stain, 200x. (c). Pleomorphic tumor cells arranged in sheets, H & E stain, 400x.
Case 8: Intra-articular synovial sarcoma of knee
A 23-years-young male presented with left knee pain following trauma for 7 months. Magnetic resonance imaging of the left knee suggested an intra-articular ganglion cyst. Curettage of the lesion yielded four ml volume of multiple gray white and pearly white soft tissue bits. Microscopic examination revealed fragments of tumor tissue consisting of an epithelial component in the form of glands lined by cuboidal cells surrounded by short and long fascicles of moderately pleomorphic spindle cells. In addition, hypocellular areas with hyalinization and calcification were observed. A diagnosis of synovial sarcoma was thought and subsequently confirmed by immunohistochemistry using CD99 and TLE1. (Figure 7). Photomicrograph of intraarticular synovial sarcoma. (a). Classical biphasic morphology showing glands surrounded by spindle cells, H & E stain, 100x. (b). Spindle-shaped cells arranged in fascicles, H & E stain, 200x. (c). Transducin-like enhancer of split 1 (TLE1) positivity in both epithelial and stromal components on immunohistochemistry, 400x.
Kidney tumors
Case 9: Ewing sarcoma of kidney
A 35-year-old female presented with a lump in the abdomen for 1 month, which was found to be a mass lesion in the left kidney on magnetic resonance imaging. Multiple hyperintense lesions in the vertebrae were also observed. Radical nephrectomy was performed with a clinical diagnosis of renal cell carcinoma with vertebral metastases. A gray-white fleshy tumor occupying the entire kidney was found on gross examination. Histopathology revealed sheets of monotonous population of small round cells with high mitotic activity and focal rosette formation. The tumor infiltrated the adjacent perinephric fat and the ureter. Immunohistochemistry was done with a broad diagnosis of small round cell tumor. Vimentin, CD45 and EMA were negative excluding Wilms’ tumor and non-Hodgkin lymphoma. CD99 showed diffuse membrane positivity, and neuron specific enolase demonstrated cytoplasmic positivity, confirming the diagnosis of Ewing sarcoma of the kidney (Figure 8). Molecular study was suggested but not done as the patient could not afford. Photomicrograph of Ewing sarcoma of the kidney. (a). Cellular tumor composed of sheets of undifferentiated small round cells, H & E stain, 400x. (b). Strong cytoplasmic positivity for neuron specific enolase, 400x. (c). Diffuse membrane positivity for CD99, 400x.
Breast tumors
Case 10: Primary plasmacytoma of breast
A 60-year-old, otherwise healthy female presented with a complaint of a lump in the left breast for 6 months. Unlike other breast tumors, the lesion involved the entire left breast. The FNAC smears predominantly showed discretely arranged small, relatively uniform cells with eccentric nuclei, which were misinterpreted as lobular carcinoma [Figure 9(a)]. Modified radical mastectomy confirmed the clinical findings, showing a diffuse and firm gray-white tumor occupying all four quadrants of the breast. Microscopically, sheets of mature and immature plasma cells were observed, which were positive for CD 138 and negative for CK, confirming the diagnosis of plasmacytoma and excluding lobular carcinoma [Figure 9(b)]. Bone marrow examination findings were unremarkable. Photomicrograph of primary plasmacytoma of breast. (a). Cytology smear showing discrete plasma cells and some binucleate forms, H & E stain, 200x. (b). Histopathology showing sheets of atypical plasma cells, H & E stain, 400x.
Discussion
Head and neck tumors
Case 1: Primary nasal meningioma
Primary extracranial meningiomas are uncommon, constituting less than 2% of all meningiomas. The head and neck region is the most common extracranial location, where it occurs in the orbit, scalp, petrous pyramid, and sinonasal tract. Meningiomas in this region are classified as primary and secondary based on the presence or absence of an intracranial tumor. Primary meningiomas of the nasal cavity are extremely rare, with an incidence of 0.1% of all sinonasal tract tumors.4,5 Extracranial meningiomas are thought to arise from arachnoid cells within the nerve sheaths, as they traverse through skull foramina or from displaced and entrapped pacchionian bodies during embryonic development.4,5 Although histologically similar to intracranial meningiomas, these tumors often present diagnostic difficulties owing to their unusual location. Primary nasal meningioma must be differentiated from other sinonasal tract tumors such as carcinoma, melanoma, lymphoma, and olfactory neuroblastoma.4,5 The diagnosis was based on the presence of whorling, psammoma bodies, uniform cells, intranuclear inclusions, strong vimentin and EMA positivity, and CK negativity. Carcinoma typically shows CK-positive pleomorphic epithelial cells, while melanoma shows epithelial and spindle cells with prominent nucleoli, intracytoplasmic melanin, and positive staining with S100, HMB45, and Melan A. Olfactory neuroblastoma and lymphoma display small round undifferentiated morphology and specific IHC markers. Accurate diagnosis requires correlation of histopathology with imaging, in which IHC plays a crucial role. Follow-up of the patient revealed multiple recurrences.
Case 2: Malignant melanoma of lacrimal sac
Extra-cutaneous malignant melanoma is rare, commonly occurring in the gastrointestinal tract, oral cavity, conjunctiva, choroid, and lacrimal sac. Most lacrimal sac lesions are inflammatory, and benign tumors outnumber malignant ones, which are typically squamous or transitional in nature. 6 Malignant melanoma of the lacrimal sac is extremely rare, with only 50 cases reported in the medical literature, constituting 4%–13% of lacrimal sac tumors and 0.7% of all melanomas. 7 The exact histogenesis is unknown, possibly originating from melanocytes migrating from the neural crest during embryogenesis. 7 Dysplastic nevi and a personal or family history of cutaneous melanoma are a few known risk factors. The prognosis is poor as mucosal melanomas tend to behave more aggressively than their cutaneous counterparts. The presence of intracytoplasmic melanin facilitated the diagnosis in this case. The diagnosis of lacrimal melanoma is often delayed because this lesion clinically mimics dacryocystitis.7,8
Case 3: Metastatic synovial sarcoma of thyroid
Synovial sarcoma, a biphasic malignant soft tissue tumor, typically occurs in the vicinity of joints, most commonly around the knee. Primary synovial sarcoma of the thyroid is extremely uncommon, with only 15 cases documented in the literature till date.9–11 Soft tissue synovial sarcoma frequently metastasizes, primarily to the lungs, followed by the lymph nodes, bones, and liver. Metastatic spread to the thyroid gland is extremely rare. 12 The present case resembled spindle cell variant of medullary carcinoma on both cytology and histopathology. Delicate fibrovascular septa with prominent vasculature, lack of pleomorphism and mitotic activity, and presence of amyloid, the typical features of medullary carcinoma, were not observed in the present case. Another differential diagnosis to consider is the sarcomatoid variant of anaplastic carcinoma. This tumor is characterized by the presence of pleomorphic spindle and giant cells, mitotic figures, and necrosis, and is positive for pancytokeratin and PAX8. The diagnosis of the present case was challenging at this singular and unusual location. A panel of immune markers and correlation with the patient’s previous clinical history helped us in the diagnosis. To the best of our knowledge, this is the second case of metastatic synovial sarcoma of the thyroid reported in the medical literature. Molecular testing for the SS18-SSX fusion gene confirms the diagnosis of synovial sarcoma.12,13
Case 4: Malignant teratoma of thyroid
Teratomas are germ cell tumors, consisting of all the three germ layers, usually occur in the gonads. Extragonadal teratomas are uncommon and typically occur in the midline. A cervical teratoma is considered of thyroid origin if the tumor is within the thyroid, has a connection with the thyroid, or when the thyroid gland is absent.14,15 Adult Thyroid teratomas are extremely rare and are usually malignant in nature. Fewer than 40 case reports of malignant teratoma of thyroid have been published in the literature. 16 The histopathology of thyroid teratomas is similar to that of teratomas elsewhere in the body. The presence of neuroectodermal tissue is an important clue for diagnosing teratomas in unusual locations, such as thyroid.14–16
Male genital tract tumors
Case 5: Papillary serous cystadenocarcinoma of paratestis
Ovarian-type surface epithelial tumors of the testis and paratestis are rare and are considered homologs to their more common ovarian counterparts in terms of morphology, immunohistochemistry, and ultrastructure. 17 The entire spectrum of ovarian tumors can occur, with serous tumors being the most common. 18 Fewer than 50 serous tumors have been described to date, of which borderline tumors are more common.17,19 The proposed histogenesis of these tumors is Mullerian metaplasia of the tunica vaginalis or from remnants of mesonephric rests.18,20 Papillary serous cystadenocarcinoma is an extremely uncommon paratesticular tumor, with only 18 cases reported worldwide, including two from India.18,19 This is an infiltrating tubulo-papillary tumor that predominantly occurs in the paratestis but can also be found in the testis. The presence of papillae, high nuclear grade tumor cells, and psammoma bodies are clues to the serous differentiation. CA125 levels are usually elevated and considered a specific marker. 20 It must be differentiated from more common and highly malignant tubulopapillary tumors of the paratestis, such as malignant mesothelioma, and extremely rare metastases and carcinoma of the rete testis. 20
Case 6: Leiomyoma of paratestis
A solid testicular tumor is often malignant and is usually a germ cell tumor. The scrotum, vulva, and penis are common extrauterine sites of genital leiomyoma. 21 Leiomyoma involving the testis is extremely rare, with only 17 cases reported in the medical literature to date. 22 The proposed origin of these tumors is the tunica propria of the seminiferous tubules, tunica albuginea, and blood vessels. 21 The differential diagnoses include benign paratesticular tumors such as fibroma and inflammatory myofibroblastic tumor, and leiomyosarcoma. The absence of high cellularity, mitotic activity, necrosis, and infiltrative growth differentiates leiomyoma from leiomyosarcoma. The continuity of the tumor with the smooth muscle of the tunica albuginea and adjacent blood vessels in the present case emphasizes the possible origin of leiomyoma from these structures. 21 Preoperative diagnosis can prevent unnecessary orchidectomy; hence, it should be considered in clinical and radiological evaluations.
Skin and soft tissue tumors
Case 7: Squamous cell carcinoma in an epidermoid cyst
Epidermoid cyst is a common benign cutaneous cystic lesion. However, the development of squamous cell carcinoma in an epidermoid cyst is exceptional, with an incidence of 0.011%–0.045%. 23 A literature review showed only 56 case reports of squamous cell carcinoma developing in an epidermoid cyst. 24 The proposed risk factors are chronic irritation, actinic damage, ultraviolet light exposure, and human papillomavirus infection. This entity must be differentiated from the more common cystic squamous cell carcinoma, which is a conventional squamous cell carcinoma with cystic change. In the present case, the cyst lined with dysplastic keratinized stratified squamous epithelium, and the continuity between the dysplastic lining epithelium and the underlying infiltrative growth confirmed the diagnosis. Malignant transformation should be suspected in large epidermoid cysts with rapid growth, ulceration of the skin or fistula formation. 25 A high index of suspicion is necessary for preoperative diagnosis to guide proper management of this condition.
Case 8: Intra-articular synovial sarcoma of knee
Primary intra-articular synovial sarcoma is extremely rare, with only 22 cases documented in the medical literature. 26 Less than 5% of all synovial sarcomas are intra-auricular, majority involving knee joint.26–28 MRI findings are often nonspecific and mimic more common benign lesions, such as villonodular synovitis, synovial chondromatosis, and loose bodies in the joint. 29 Microscopic features are similar to those of extra-articular synovial sarcoma, with the majority of cases exhibiting classical biphasic morphology. The presence of bimodal morphology with calcification combined with IHC helped us in making the correct diagnosis. Molecular studies are essential for confirming this diagnosis.
Kidney tumor
Case 9: Ewing sarcoma of kidney
Ewing sarcoma, a common malignant tumor of bone, belongs to the Ewing family of tumors. It is characterized by a mutated EWSR1 gene, which is most often due to t (11; 22) (q24; 12). These tumors are poorly differentiated, cellular round cell sarcomas with variable neural differentiation in the form of Homer-Wright rosettes and neuropil.30,31 Extra-skeletal Ewing sarcoma can occur anywhere in the body, with the most common site being the soft tissue of the extremities. Renal Ewing sarcoma is an aggressive tumor in young patients, with an advanced stage of presentation and poor prognosis. The cell of origin could be embryonic neural cells of the kidney. This tumor shares morphological resemblances with other small round cell tumors, such as blastemic adult Wilm tumor, neuroblastoma, non-Hodgkin lymphoma, and synovial sarcoma. A combination of immunohistochemical markers, including CD 99, FLI-1, NSE, and vimentin, is necessary for an accurate diagnosis, along with cytogenetic analysis for confirmation. The possibility of Ewing sarcoma should be considered in the differential diagnosis, when a small round cell tumor with an aggressive course occurs in the kidney of a young patient.30,31
Breast tumor
Case 10: Primary plasmacytoma of breast
Primary plasmacytoma involving the breast is extremely rare. Plasmacytoma of breast occurs secondarily in cases of multiple myeloma or as a part of multifocal myeloma or exceptionally rarely as an isolated primary plasmacytoma. 32 A total of 45 cases of primary plasmacytoma of the breast have been reported in the literature. 33 Accurate preoperative diagnosis of plasmacytoma, although difficult, is vital to prevent unnecessary mastectomy. FNAC is a useful tool for the initial diagnosis, particularly in known cases of myeloma. The differential diagnoses to be considered are plasma cell mastitis, MALT lymphoma and lobular carcinoma. Confirmation of plasmacytoma requires CD 138, kappa, and lambda light chain positivity and CK negativity. 32 The presence of polyclonal plasma cells and mixed inflammatory cells is a feature of plasma cell mastitis. Whereas, lobular carcinoma shows discrete small uniform cells with intracytoplasmic vacuoles. MALT lymphoma is a low grade B cell neoplasm that is positive for specific B cell markers. Histopathology and immunohistochemistry are the gold standards for diagnosing primary plasmacytoma of breast. Additional diagnostic criteria include normal bone marrow findings, a negative skeletal survey, and the absence of end-organ damage. 34
The summary of the diagnostic features of the tumors.

Conclusion
These tumors are extremely rare, making preoperative diagnosis difficult. As a group, these tumors are less studied, and with limited experience and available literature, they are easily misinterpreted, resulting in serious mismanagement of patients. The lack of knowledge regarding pathogenic mechanisms and medical expertise highlights the need for more research in this field.
The current study is limited by small sample size, limited IHC panel, and in some cases, lack of molecular confirmation due to financial limitations and unavailability.
In conclusion, tumors at unusual locations are interesting to explore, challenging to solve, and require awareness and a high degree of suspicion for diagnosis. Accurate diagnosis requires multidisciplinary approach, as well as immunohistochemistry and molecular genetics for confirmation.
Footnotes
Acknowledgements
We would like to thank all our patients for their co-operation.
Ethical approval
Ethical approval to report these cases was obtained from “Rangaraya Medical college institutional ethics committee” (approval number- IEC/RMC/2018/379).
Consent for publication
Written informed consent was obtained from all patients for the publication of their anonymized clinical information and accompanying gross and histopathology images in this article. A copy of the written consent is available for review by the Editor-in-Chief upon request.
Author Contribution
Dr. Rajyalakshmi Rallapalli conceptualized the study, collected the data and wrote the original draft. Dr Kusa Raju Pyla collected the data, contributed to the intellectual content and edited the manuscript. Dr Beulah priscilla Maddirala did the critical corrections and revised the manuscript. Dr Kishore Kumar Ch analysed the data collected and edited the manuscript. Dr Ganesh Basina analysed the data and did language editing. Dr. Rajyalakshmi Rallapalli has written the first draft of the article. All authors reviewed and edited the manuscript and approved the final version of the manuscript.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data generated and analyzed during this study, including patient complaints, surgical details, and histopathology and immunohistochemistry data, are not publicly available due to ethical and privacy restrictions to protect patient confidentiality. These data are recorded in departmental registers and are available from the corresponding author upon reasonable request, subject to approval by the institutional ethics committee.