
Abstract
Soft tissue sarcomas are rarely associated with mutations of the MEN1 gene. We report a patient with a large retroperitoneal pleomorphic liposarcoma harboring a rare mutation of the MEN1 gene not previously reported to be associated with soft tissue sarcomas. This report expands the known spectrum of MEN1-associated cancers.
Case report
A 41-year-old previously healthy man presented to our hospital with a 24-h history of right abdominal and flank pain. His physical examination was unremarkable except for hypertension and tenderness in the right upper quadrant and right flank. Workup including CT imaging identified two large tumors involving the right adrenal gland and right hepatic lobe (Figure 1(a)–(c)), with some contiguity between the masses. The liver mass was associated with an acute-appearing subcapsular hematoma in the liver, which likely contributed to his pain symptoms. His medical history was pertinent for nephrolithiasis and hypertension. On laboratory workup, he was also found to have hypercalcemia (serum calcium 11.7 mg/dL) and hyperparathyroidism (serum parathyroid hormone 181 pg/mL). His presentation was initially concerning for malignant pheochromocytoma based on its location and his associated history, and therefore a diagnostic evaluation for a functional hormone-secreting adrenal tumor and adrenal-protocol imaging were completed. Laboratory testing was consistent with the presence of a cortisol-secreting tumor (urine free cortisol 346.6 µg/dL), and two dexamethasone suppression tests did not appropriately suppress his serum cortisol (cortisol after 1 mg dexamethasone suppression test, 10.1 µg/dL; cortisol after 2 mg dexamethasone suppression test, 9.1 µg/dL). Plasma metanephrines (0.64 nmol/L) and normetanephrines (0.64 nmol/L) were only mildly elevated and therefore not suggestive of a primary locally advanced pheochromocytoma. MR imaging demonstrated that the masses were not consistent with classical features of adrenocortical carcinoma, hepatocellular carcinoma, or cholangiocarcinoma, and no other distant sites of disease were identified on staging imaging including CT chest. 131I-metaiodobenzylguanidine imaging did not clearly demonstrate scintigraphic findings of a pheochromocytoma. (a)-(c) cross-sectional views of retroperitoneal tumor involving the right hepatic lobe and right adrenal gland, with associated liver hematoma.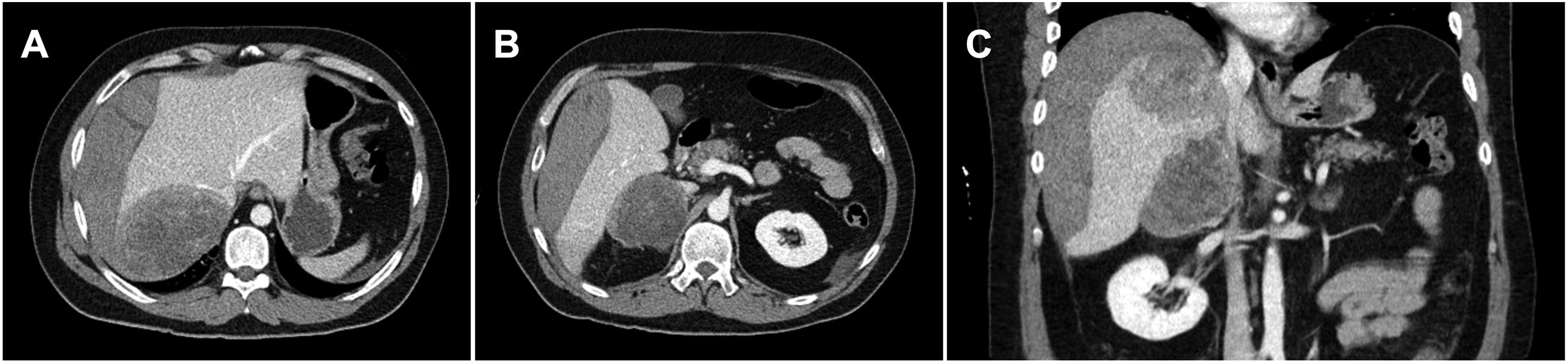
Despite this extensive evaluation, there was still uncertainty regarding the primary etiology of the masses. In particular, metastatic adrenocortical carcinoma to the liver, metastatic primary liver cancer to the adrenal gland, or a primary retroperitoneal tumor were still diagnostic possibilities, each with potentially divergent management options. Given this uncertainty, a CT-guided percutaneous biopsy of the adrenal mass was performed, which demonstrated that this tumor was most consistent with a pleomorphic malignant neoplasm with vacuolated cells, with consideration of a tumor of adipocytic origin. Although FISH did not demonstrate MDM2 gene amplification, the possibility of a lipomatous tumor was not excluded.
Immunohistochemical stains identified tumor cells diffusely positive for GATA3 and p53, with patchy staining for cytokeratin oscar, glypican-3, WT-1, CAIX (weak), PAX8, and CD31. There were rare cells staining for pan-cytokeratin and arginase. The cells were negative for AE1/AE3, keratin 7, keratin 20, hepatocyte antigen, RCC, inhibin, synaptophysin, chromogranin, S100, TTF-1, SOX-10, HMB-45, uroplakin, CD30, CAM5.2, desmin, Oct3/4, CD117, CD45, smooth muscle actin, calretinin, SS18-SSX, and CDX2. Non-specific focal staining was seen for AFP, DOG-1, and ERG. BAP-1 nuclear staining was retained.
As a locally advanced primary retroperitoneal tumor was identified, he underwent open right hepatic lobectomy with cholecystectomy and en bloc right adrenalectomy with complete extirpation of the contiguous tumors. Pathology confirmed a retroperitoneal high-grade pleomorphic liposarcoma involving the right adrenal gland and extending into the right lobe of the liver (Figure 2(a)), measuring 21 cm in size and with negative margins. The final pathologic stage was T4N0. There was 30%–40% tumor necrosis present, no lymphovascular invasion identified, and 30 mitoses per 10 high-power fields (Figure 2(b)–(c)). The specimen stained positive for vimentin only, but was negative for S100, desmin, SMA, hepatocyte antigen, arginase, HMB45, MiTF, caldesmon, and calponin. FISH testing for MDM2 amplification was again negative. (a) gross image of resected tumor. (b) histopathology at 100× power. (c) histopathology at 400× power.
His postoperative course was complicated by a controlled but persistent bile leak requiring percutaneous drain placement as well as ERCP with sphincterotomy and biliary stent placement. He unfortunately developed early recurrence of disease involving a mass in the caudate lobe of the liver, as well as eventual right upper quadrant peritoneal sarcomatosis and diffuse skeletal metastasis with cauda equina syndrome. He underwent palliative spine and sacrum radiation and was treated with doxorubicin, ifosfamide, and mesna (AIM), which were discontinued due to disease progression and regimen intolerance, followed by courses of eribulin and trabectedin, which were each discontinued due to disease progression, and later palbociclib.
In subsequent evaluation of hyperparathyroidism and nephrolithiasis, an ultrasound of the thyroid gland identified an 8-mm parathyroid adenoma candidate near the inferior aspect of the right thyroid lobe. Given the need for multimodal therapies, he is maintained on calcimimetics and has not undergone parathyroidectomy.
His personal and family history was concerning for a potential germline mutation, as his brother had primary hyperparathyroidism; his sister had an unknown lung cancer and died from this at age 42; another sister had a pituitary adenoma, hyperthyroidism, and hypercalcemia; his father died at age 45 from bleeding stomach ulcers and had multiple skin tags; his paternal uncle had stomach cancer at age 70; and one maternal aunt had breast cancer in her 60s. Another sister has Hashimoto’s disease, and her daughter had Hashimoto’s disease as well. His paternal uncles had multiple skin tags. His mother’s family history was unremarkable for cancer.
He underwent 67-gene germline panel testing that revealed a heterozygous pathogenic mutation in exon one of the MEN1 gene, variant c.2T>A (M1K), confirming multiple endocrine neoplasia type 1 (MEN1) syndrome. Next-generation sequencing performed on the resected tumor identified this MEN1 M1K mutation, a TP53 V127G mutation, a GNAS R201C mutation, and CCNE1 amplification. Immunohistochemical staining to confirm loss of menin expression was not performed at the time of pathologic evaluation, as MEN1 was not included as a possible underlying etiology at the time. The tumor was microsatellite stable and had a tumor mutational burden of one mutation per megabase pair. He subsequently underwent additional screening with MR brain, which did not demonstrate a pituitary mass. No pancreatic masses had been noted on MR imaging.
The MEN1 M1K mutation disrupts the initiator methionine residue of MEN1 mRNA. While there are 1012 unique mutations of the MEN1 gene reported in the Catalogue of Somatic Mutations in Cancer (COSMIC) Database,1,2 this particular mutation has not been described. No MEN1 mutations occur in any liposarcoma included in the COSMIC database. cBioPortal3,4 was screened for soft tissue sarcomas harboring MEN1 alterations. The overall MEN1 mutation frequency was 0.4%, with seven putative driver alterations across five datasets including 3071 samples.5–8 One pleomorphic liposarcoma was found to harbor a MEN1 I85Sfs*33 frameshift mutation. 6 No MEN1 M1K mutations in any cancer type have been reported in the cBioPortal database.
CCNE1 encodes the protein cyclin E158, and its amplification leads to overexpression of this oncogene involved in cell cycle regulation. CCNE1 amplification is found in 33% of pleomorphic liposarcomas and 2% of dedifferentiated liposarcomas. 7 Additionally, CCNE1 amplification has been identified as a driver alteration in uterine leiomyosarcoma and has been noted in a number of other solid malignancies, including adenocarcinomas. 9 Loss-of-function mutations of the TP53 tumor suppressor protein are common in advanced cancers and have been found in 17% of pleomorphic liposarcomas. 7 GNAS mutations are rarely associated with sarcoma.
Discussion
MEN1 syndrome is an autosomal dominant hereditary condition that is most commonly associated with endocrine tumors of the parathyroid glands, pancreas, and pituitary gland. The prevalence of MEN1 is one in 30,000 people. The MEN1 gene encodes the menin protein, which acts as a tumor suppressor in the regulation of cell cycle, gene expression, and DNA repair pathways. Pathogenic mutations in the MEN1 gene are associated with MEN1 syndrome and familial isolated hyperparathyroidism. The gene is also related to thyroid cancers and paraganglioma-pheochromocytoma syndrome. MEN1 alterations are implicated in multiple cancer types, including many distinct from a neuroendocrine origin. 10 An association with MEN1 mutations has also been reported for angiofibromas, collagenomas, lipomas, meningiomas, ependymomas, and leiomyomas.
Soft tissue sarcomas are rarely associated with mutations of the MEN1 gene. One case of bone sarcoma was identified to harbor the MEN1 c.2T>A mutation reported here and co-occurred with a putative oncogenic mutation in EXT1. 11 Another case of soft tissue sarcoma in the setting of a different MEN1 gene mutation has been reported in the literature. 12 There are few MEN1 mutations in patients with soft tissue sarcoma reported in cBioPortal.
There are several indications to conduct germline testing for patients suspected to have MEN1, which may include personal history of two or more MEN1-associated tumors, family history of MEN1 in a first-degree relative, atypical phenotypes of MEN1 such as parathyroid hyperplasia or adenomata younger than age 40, recurrent or multigland hyperparathyroidism, familial isolated hyperparathyroidism, gastrinoma, or multifocal gastro-entero-pancreatic neuroendocrine tumors. Screening patients with soft tissue sarcomas would likely confer an extremely low yield of MEN1 identification, based on the rarity of their association as highlighted here, and therefore would likely not be cost-effective for this purpose.
While this case of a single patient cannot guide all patients with MEN1 or with soft tissue sarcomas, this report highlights a novel association between the two, which may inform management of similar patients with soft tissue sarcomas and potentially expand the known clinical spectrum of MEN1.
Conclusion
We report a patient with a large retroperitoneal pleomorphic liposarcoma harboring a rare mutation of the MEN1 gene not previously reported to be associated with soft tissue sarcomas. Soft tissue sarcomas in the setting of MEN1 syndrome are extremely rare. This report expands the known spectrum of MEN1-associated cancers.
Footnotes
Acknowledgements
None
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Contributorship
BDF wrote the first draft of the manuscript. All authors reviewed and edited the manuscript and approved the final version of the manuscript.
Ethical statement
Data Availability Statement
Data availability is not applicable because there are no primary data reported in our manuscript.