
Abstract
Diffuse hemispheric glioma, H3 G34-mutant, is a recently recognized distinct high-grade glioma with a dismal prognosis. In addition to the H3 G34 missense mutation, numerous genetic events have been identified in these malignant tumors, including ATRX, TP53, and, rarely, BRAF genes. There are only a few reports to date that have identified BRAF mutations in diffuse hemispheric glioma, H3 G34-mutant. Moreover, to our knowledge, gains of the BRAF locus have yet to be described. Here, we present a case of an 11-year-old male with a diffuse hemispheric glioma, H3 G34-mutant, found to have novel gains of the BRAF locus. Furthermore, we emphasize the current genetic landscape of diffuse hemispheric glioma, H3 G34-mutant, and implications of an aberrant BRAF signaling pathway.
Introduction
In the latest World Health Organization (WHO) Classification of Tumors of the Central Nervous System, diffuse hemispheric glioma, H3 G34-mutant, is recognized as a distinct high-grade glioma 1 with a dismal overall survival.2–9 Genetic events (e.g. single nucleotide variants, gene loss, rearrangements, or amplifications) have been reported in numerous genes in diffuse hemispheric glioma, H3 G34-mutant: the most notable of which include the pathognomonic H3 G34R or H3 G34V missense mutations.2–9 However, the only additional genetic events found to influence overall survival identified thus far include PDGFRA and EGFR amplifications. 2 Additional genetic events in the BRAF gene (i.e. single nucleotide variants, gene arrangements) have been reported in a few cases of diffuse hemispheric glioma, H3 G34-mutant. 2 However, gains of the BRAF locus have yet to be reported. Here, we present the first report of a diffuse hemispheric glioma, H3 G34-mutant with gains of the BRAF locus identified in a pediatric patient.
Case illustration
An 11-year-old male presented to the emergency department with 2 weeks of progressively worsening headaches and gait imbalance. Neurologic examination was unremarkable. Past medical history was unremarkable. Magnetic resonance imaging (MRI) of the brain demonstrated a well-circumscribed large heterogeneously enhancing lesion of the right parietotemporal lobe with a cystic component without significant surrounding vasogenic edema (Figure 1). Following an interdisciplinary discussion with pediatric oncology and radiation oncology, dexamethasone was initiated and neurosurgical intervention was recommended. Pre-operative MRI demonstrates a large right parietotemporal heterogeneously enhancing lesion with a cystic component and hypervascularity around the periphery. (a)–(c) T1-weighted images with contrast. (d, e) T2-weighted images. (f) Perfusion image.
A stereotactic right craniotomy was performed for resection of the right parietotemporal lesion. Gross total resection was achieved as post-operative MRI was without evidence of residual tumor. The patient tolerated the procedure well and without complication. The patient subsequently underwent proton-beam radiotherapy; however, chemotherapy was not recommended by pediatric oncology given gross-total resection of the tumor. Histopathology demonstrated a mixture of morphologic patterns. Much of the tumor had monotonous and slightly spindled cells with a high proliferative index, numerous mitotic figures, some perivascular pseudorosettes as well as areas of true rosette formation suggestive of ependymal morphology. Other areas were more astrocytic in appearance with complex microvascular change as well as areas of extensive geographic and palisading necrosis. There appeared to be minimal infiltration into the surrounding benign brain parenchyma. Some sections showed numerous overtly anaplastic and pleomorphic giant cells with enlarged hyperchromatic nuclei and multinucleation. Overall, these findings were histologically consistent with high-grade glioma (Figure 2). GFAP was positive mostly highlighting astrocytic processes between cells with variable cellular positivity. Synaptophysin exhibited variable positivity and intensity. p53 exhibited diffuse strong positivity in viable tumor consistent with a p53 mutation. Ki-67 was significantly elevated with approximately 60% overall with some areas approaching 80%. ATRX exhibited loss of nuclear staining. H3 G34R/V exhibited variable positivity with much of the tumor demonstrating weak nuclear staining in approximately 10% but some areas with dense nuclear staining in clusters (Figure 3). Additional immunohistochemical stains including cytokeratin AE1/AE3, CAM5.2, EMA, chromogranin, S100, IDH1 R132H, OLIG2, L1CAM, and H3 K27M were negative. Histopathology consistent with high-grade glioma with variable cellular morphology, a high proliferative index, numerous mitotic figures, perivascular pseudorosettes, true rosettes, complex microvascular changes, extensive geographic and palisading necrosis, interspersed entrapped neurons as well as some malignant multinucleated cells. (a, b) Perivascular rosettes (10x). (c, d) Perivascular rosettes (20x). (e) Embryonal area (5x). (f) Embryonal area (10x). (g) Embryonal area with rosettes (20x). (h) Embryonal immature area (40x). (i) Mix of morphologies with ganglion cells (20x). (j) Embryonal immature area with fascicular architecture and high-grade nuclei (20x). (k) Giant cell area with areas of necrosis (10x). (l) Giant cell area with areas of necrosis (20x). Immunohistochemistry consistent with diffuse hemispheric glioma, H3 G34-mutant. (a)–(c) H3 G34R/V stain with variable positivity with much of the tumor exhibiting weak nuclear staining in approximately 10% but some areas with dense 3 + nuclear staining in clusters (10x). (d) H3 K27M negative stain (10x). (e) ATRX with loss of nuclear staining (10x). (f) IDH1 R132H negative stain (10x). (g) OLIG2 negative stain (10x). (h) p53 mutated, strong positivity in viable tumor (10x). (i) Ki-67 significantly elevated with approximately 60% overall with some areas approaching 80%.

A NeoTYPE brain tumor profile demonstrated several single nucleotide variants, including ATRX Q289Vfs*6, H3-3A G34R, and TP53 N131del. There was no evidence of RNA fusion, EGFRvIII mutation, or MGMT promoter methylation. Additionally, no abnormalities were detected in HIST1H3C, IDH1, or IDH2. Fluorescent in situ hybridization did not detect PDGFRA amplification, 1p/19q co-deletion, BRAF gene rearrangement, MET amplification, MYC/N amplification, or PTEN deletion. However, gains of the BRAF locus (>2F, 44%) were observed indicative of gains or extra copies of the BRAF gene region on chromosome seven (Figure 4). Indices for significant gains of BRAF were noted as follows: negative <10%, borderline positive 10–20%, positive >20%. Methylation profiling indicated the gains in the BRAF region on chromosome seven but did not show gains of the entire chromosome seven or the region encoding EGFR. As such, a diagnosis of diffuse hemispheric glioma, H3 G34-mutant, was established and noted to have gains of the BRAF locus. Florescence in situ hybridization. (a) 19q deletion not detected. (b) 1p deletion not detected. (c) BRAF with abnormal signal pattern of >2F (44%) consistent with gains or extra copies of the BRAF locus. (d) MET non-amplified. (e) MYCN non-amplified. (f) PDGFRA non-amplified.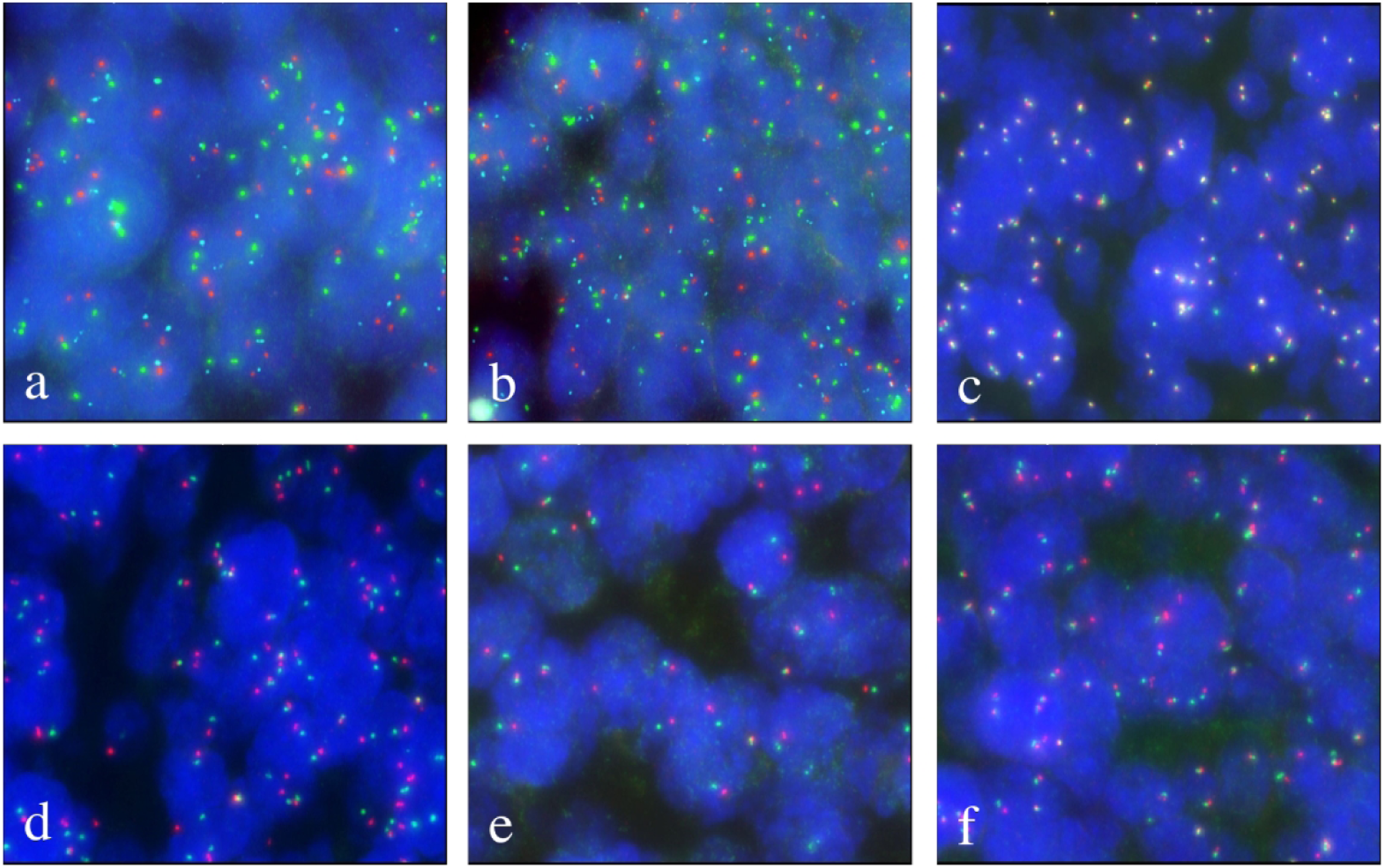
Discussion
Somatic gain-of-function mutations in the genes that encode histone H3 proteins characterize two distinct types of high-grade gliomas including diffuse midline glioma H3 K27-mutant and diffuse hemispheric glioma, H3 G34-mutant.1,10 These tumors exhibit mutations in several H3 genes, including H3.1, H3.2, and, most commonly, H3.3. A lysine to methionine substitution at codon 27 in the HIST1H3B/C or H3F3A genes (H3 K27M-mutant) is pathognomonic for an aggressive grade IV high-grade midline glioma, known as diffuse midline glioma H3 K27-mutant, which carries a poor prognosis.1,2 Comparatively, glycine to valine or arginine substitutions at codon 34 in the H3F3A gene (H3 G34-mutant) characterize an additional grade IV high-grade glioma, known as diffuse hemispheric glioma, H3 G34-mutant, which primarily occurs off midline in the cerebral hemispheres.1,2 Prior studies have shown that the H3 G34V exhibits a worse prognosis compared to the H3 G34R variant with a median overall survival of 9.9 vs 14.8 months, respectively. 2 Although the H3 K27 and H3 G34 mutations are pathognomonic for their respective diagnoses, the molecular mechanisms by which these histone mutations contribute to tumorigenesis remain to be elucidated.1,2,8
In the largest study to date, Vuong et al. evaluated the prognostic significance of genetic events in 257 diffuse hemispheric gliomas, H3 G34-mutant, compromised within 20 studies. 2 Overall, the authors found numerous genetic events within these tumors including 10q loss (42.9%), ATRX loss (83.3%), ATRX mutations (87.5%), BRAF mutations (1.9%), CDKN2A/B deletion (22.0%), EGFR amplification (7.5%), IDH mutations (0.8%), MET amplification (4.6%), MGMT methylation (79.5%), MYC/N amplification (8.9%), NF1 mutations (13.4%), PDGFRA amplification (16.4%), PDGFRA mutations (47.3%), PIK3CA mutations (1.4%), PTEN mutations (11.8%), TERT mutations (1.4%), and TP53 mutations (94.9%). 2 However, only PDGFRA and EGFR amplifications were found to negatively influence overall survival in addition to the G34V genotype. 2 Importantly, the authors found only two mutations (1.9%) within the BRAF gene. 2 However, there was no report of any gains or extra copies of the BRAF gene region in their integrated analysis. 2 In another study by Picart et al., independent molecular analysis of 17 patients revealed ATRX loss (92.9%), EGFR amplification (14.3%), MGMT methylation (81.8%), and TP53 mutations (87.5%). 4 There was no evidence of mutations in IDH, PTEN, or BRAF genes in the cohort. 4 Interestingly, Wood et al. reported a case of post-treatment hypermutation with acquired missense mutations in mismatch repair (MMR) proteins, MSH6 and MSH2. 5 Similar to prior studies, the case we present herein exhibited common genetic events, including ATRX loss and TP53 mutations, in addition to novel gains of the BRAF locus. Although only PDGFRA and EGFR amplifications have been shown to influence overall survival, these studies highlight the mutational diversity of diffuse hemispheric gliomas, H3-G34 mutant.2–9
Several central nervous system tumors have been discovered to exhibit BRAF single nucleotide variants (e.g. BRAFV600E) or gene rearrangements (e.g. KIAA1549-BRAF), including glial tumors (e.g. pilocytic astrocytoma, pleomorphic xanthoastrocytoma, anaplastic astrocytoma) and glioneuronal tumors (e.g. ganglioglioma, desmoplastic infantile astrocytoma, dysembroplastic neuroepithelial tumor).11,12 Similarly, BRAF gains have been reported in pilocytic astrocytoma, oligodendroglioma, and glioblastoma.13,14 However, the clinical significance of BRAF gains is currently not known.13,14 As tumoral genotyping becomes commonplace, it necessitates clinicians to acquire and maintain a keen understanding of the oncologic signaling pathways in order to understand how such genetic events drive tumorigenesis and how potential therapeutics may target these aberrant signaling pathways.
The mitogen-activated protein kinase (MAPK) kinase/extracellular signal-regulated kinase (ERK) pathway regulates numerous biological functions, such as cell proliferation, differentiation, senescence, and survival.12,15,16 The MAP/ERK kinase (MEK) is the most potent activator of v-raf murine sarcoma viral oncogene homolog B1 (BRAF).15,16 Mutations in the RAS/BRAF/MEK/ERK pathway and BRAF gene occur in approximately 30% and 7% of cancers, respectively.
16
The RAS/BRAF/MEK/ERK pathway serves as a signal transduction pathway between the extracellular surroundings and the nucleus (Figure 5).15,16 Several extracellular signals, such as cytokines, hormones, and growth factors, interact with their receptors to activate the G-protein of the RAS family.15,16 Activated BRAF signals downstream via MEK to active ERK, which eventually activates transcriptional factors to influence numerous biochemical processes, such as cell proliferation.15,16 Early animal models with Ras-1 induced glioma and blockade of the BRAF hotspot mutation in brain tumor cell cultures demonstrated that tumor growth is regulated via the MAPK/ERK signaling pathway as depicted in tumors outside the bounds of the blood-brain barrier, such as melanoma.17,18 Remarkably, activation of BRAF in human neural stem cells and progenitor cells leads to tumoral growth as well as oncogene-induced senescence in several low-grade brain tumors.
19
Behling et al. postulated that this may explain the relatively high frequency of BRAF mutated brain tumors associated with favorable clinical outcomes.
12
In an elegant study by Robinson and colleagues., the authors demonstrated that BRAF mutation with Akt activation or Ink4A/ARF loss is necessary to create brain tumors with high-grade appearance.
20
Given the role BRAF may play in potential tumorigenesis, there is an emphasis placed on testing for BRAF mutations or fusions as targeted therapeutics for BRAF alternations. Numerous clinical trials are currently ongoing investigating the application of various targeted therapeutics in gliomas exhibiting BRAF aberrations (NCT01748149,
21
NCT04201457,
22
NCT03973918
23
). Schematic depiction of the RAS/BRAF/MEK/ERK pathway. Several extracellular signals interact with their receptors to activate the G-protein of the RAS family. Activated BRAF signals downstream via MEK to activate ERK, which eventually activates transcriptional factors to influence numerous biochemical processes. Abbreviations: GFR: growth factor receptor.
Conclusions
Here we describe the first report of a diffuse hemispheric glioma, H3 G34-mutant, to exhibit gains of the BRAF locus. Further studies are necessary to elucidate the clinical significance of BRAF gains in diffuse hemispheric glioma, H3 G34-mutant.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases.
Informed consent
Written informed consent was obtained from the legally authorized representative for the patient’s anonymized information to be published in this article.