
Abstract
Pineal apoplexy is a rare clinical condition. Its common symptoms include headaches, nausea, vomiting, ataxia, and gaze paralysis. These symptoms are mainly caused by obstructive hydrocephalus or direct compression of the cerebellum or midbrain. There have been no previous reports on the development of a recurrent pineal parenchymal tumor of intermediate differentiation (PPTID) with intratumoral hemorrhage. We report a case of PPTID with intratumoral hemorrhage. A 44-year-old woman developed recurrent PPTID following tumor removal and ventriculoperitoneal shunting in 2010. She visited the emergency department in April 2021 for sudden-onset dizziness and generalized weakness. Blurring of vision occurred and progressed over the previous month. Neurological examination revealed upward conjugate gaze paralysis. Brain computed tomography revealed a hyperdense lesion in the pineal region, and a recurrent tumor with hemorrhage was suspected. Magnetic resonance imaging of the brain confirmed a pineal tumor with intratumoral hemorrhage. The pineal tumor and hematoma were surgically removed via the suboccipital transtentorial approach. The patient was discharged from the hospital 2 weeks after the surgery. The pathological findings were consistent with the diagnosis of recurrent PPTID. PPTID is a rare tumor, accounting for less than 0.1% of primary central nervous system tumors. Pineal apoplexy is rare, and its incidence and clinical significance remain unclear. There have only been nine reported cases of pineal apoplexy, associated with pineal parenchymal tumors. The recurrence of PPTID with apoplectic hemorrhage after 10 years has not been reported. Despite its rarity, PPTID with apoplexy should be considered in patients with PPTID who develop sudden-onset neurological symptoms.
Keywords
Introduction
Pineal parenchymal tumor of intermediate differentiation (PPTID) is a pathological classification of pineal tumors. 1 It was first classified by the World Health Organization (WHO) in 2007, and it accounts for less than 0.1% of primary central nervous system tumors. 1 PPTID affects patients of all age groups. Its incidence peaks twice: during adulthood and senescence. On brain computed tomography (CT), PPTID are lobulated, vascular pineal region masses that can extend into adjacent structures such as the ventricles or thalami. 2 The magnetic resonance imaging (MRI) features of PPTID typically show a well-defined mass with heterogeneous enhancement and areas of necrosis. On T1-weighted images, the tumor usually appears hypointense, while on T2-weighted images, it appears hyperintense. PPTID can also cause hydrocephalus, which may be visible on an MRI. 3 Pineal hemorrhagic apoplexy is a rare clinical manifestation. Moreover, there have only been nine reported cases of apoplectic pineal tumors, including the present case. 4 Its common symptoms include headaches, nausea, vomiting, ataxia, syncope, and gaze paralysis. These symptoms are mainly caused by obstructive hydrocephalus or direct compression of the adjacent structures. 3 There is currently no established standard treatment for PPTID, but achieving gross total resection is the most important factor for optimal outcomes. After surgery, radiotherapy is commonly used to eliminate any possible remaining tumor cells. The role of chemotherapy in PPTID treatment is still being studied, and its effectiveness is uncertain. 5 The clinical prognosis is reportedly related to the histopathological grade. Recurrent PPTID with intratumoral hemorrhage has not been reported. This study reports the first case of recurrent PPTID with hemorrhagic apoplexy.
Case report
A 44-year-old woman without hypertension, antiplatelet use, or anticoagulant use developed recurrent PPTID 10 years after she received tumor removal and ventriculoperitoneal shunting. In 2010, after she received total pineal tumor resection, it was recommended that she undergo postoperative radiotherapy; however, she missed the follow-up due to personal reasons. She visited our hospital’s emergency department in April 2021, presenting with sudden-onset dizziness and generalized weakness. The blurring of vision had progressed since 1 month before the visit. Neurological examination revealed upward conjugate gaze paralysis. The non-enhanced brain CT revealed a hyperdense lesion in the pineal region and lateral ventricles (Figure 1). A recurrent tumor with hemorrhage was suspected. After admission, a brain MRI confirmed a pineal tumor with hemorrhage (Figure 2). Later, the patient underwent subtotal tumor removal and hematoma removal via the suboccipital transtentorial approach. The patient was discharged 2 weeks postoperatively. The neurological examination when she was discharged showed still-existing upward conjugate gaze paralysis. A hyperdense lesion in the pineal region on non-enhanced CT examination. (A) T1-weighted axial magnetic resonance image shows a hypointense lesion in the pineal region. (B) T2-weighted axial view shows an isointense lesion with perifocal edema in the pineal region. (C) T2 fluidattenuated inversion recovery (FLAIR) axial view shows an isointense lesion with perifocal edema in the pineal region. (D) T1 axial view with contrast shows an enhanced lesion in the pineal region. (E) T1 coronal view with contrast shows an enhanced lesion in the pineal region and ventricles. (F) T1 sagittal view with contrast shows an enhanced lesion in the pineal region and ventricles.

On gross inspection, the tumor was gray and had a soft texture. Histological analysis revealed moderate tumor cells composed of small round cells, most of which had uniform nuclei, fine chromatin, and small nucleoli. Low to moderate mitotic activity and nuclear atypia were observed (Figure 3). Some small, irregular, and flower-like pineocytomatous rosettes were focally observed (Figure 4). Immunohistochemical analysis revealed that the tumor was positive for synaptophysin (Figure 5), neurofilament protein (Figure 6), and neuron-specific enolase. Meanwhile, it was negative for Sal-like protein 4, cytokeratin AE1/AE3, and leukocyte common antigen. The Ki-67 proliferation index was 12% (Figure 7). The tumor was diagnosed as a grade II PPTID according to WHO standards. Histological analysis reveals that the moderate tumor cells are composed of small round cells, most of which have uniform nuclei, fine chromatin, and small nucleoli. Low to moderate levels of mitotic activity and nuclear atypia are observed. Small irregular flower-like pineocytomatous rosettes are focally observed. The darker staining in immunohistochemistry is positive for synaptophysin. The darker staining in immunohistochemistry is positive for neurofilament protein. The Ki-67 proliferation index is 12% (observed as darker staining in this figure).




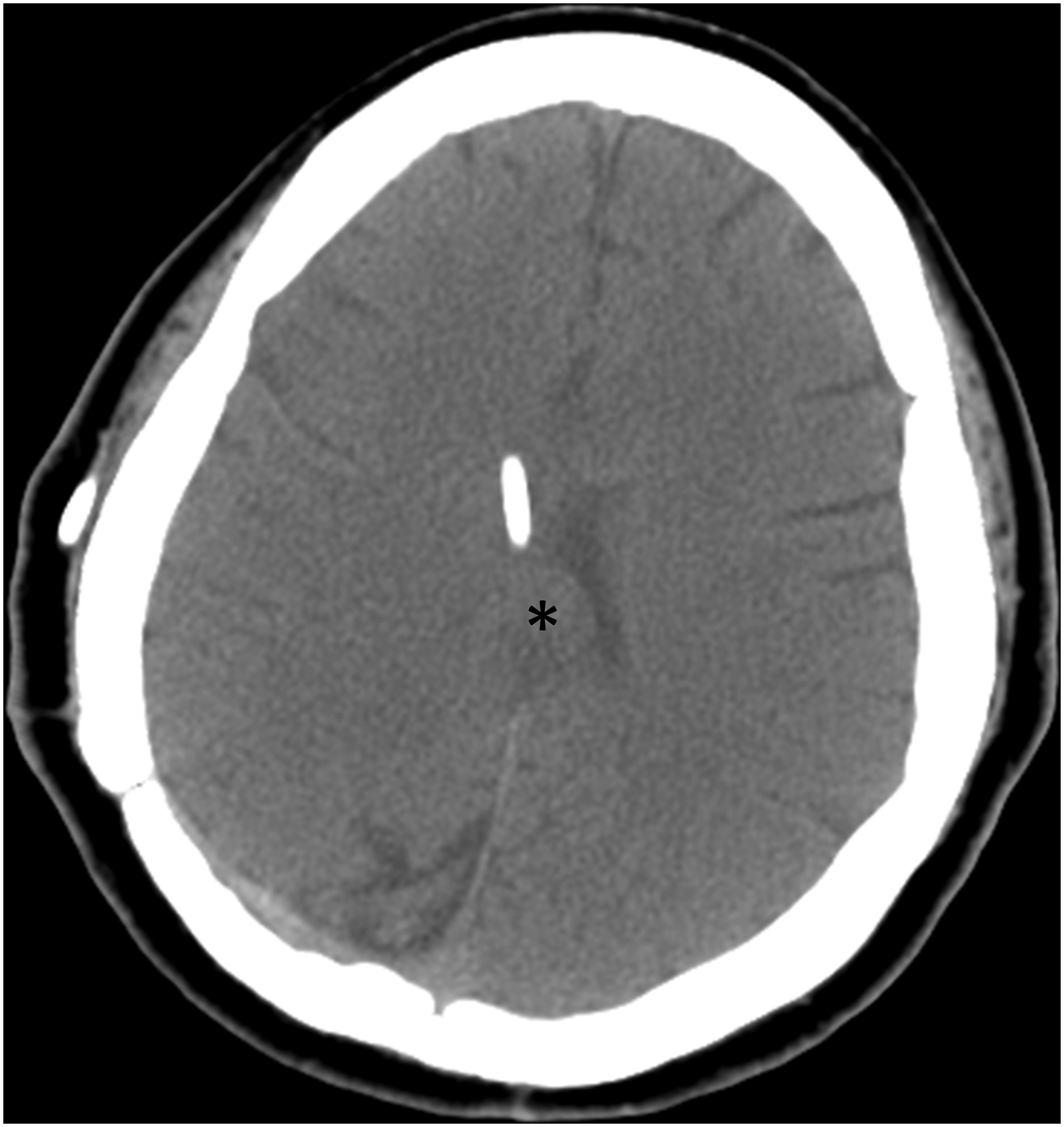
Two weeks after the subtotal tumor removal surgery, a non-enhanced brain CT scan showed a residual tumor in the pineal region and lateral ventricles (Figure 8). Although the preoperative brain MRI showed an occupied tumor in the ventricles, the spinal MRI did not provide any evidence of cerebrospinal fluid seeding. Therefore, we treated the residual PPTID with radiotherapy. Stereotactic radiotherapy (30 Gy in 10 fractions) was administered. The patient remained asymptomatic until the 1-year and 11-month follow-up visit (Figure 9). The postoperative non-enhanced CT 2 weeks after the subtotal tumor resection shows an isodense lesion (asterisk) in the pineal region. The size of the tumor was about 2.3 cm. The follow-up non-enhanced brain CT 1-year and 5-month after the subtotal tumor resection shows an isodense lesion (asterisk) in the pineal region. The size of the tumor was about 1.8 cm.
This case report was approved by the institutional review board of our hospital. A signed patient permission form was obtained from the patient.
Discussion
Pineal parenchymal tumors originate from the melatonin-producing cells of the pineal gland and account for approximately 30% of pineal region tumors. 6 These tumors vary in histopathological features, with pineocytomas being the most benign and pineoblastomas being the most malignant. PPTID falls between these two ends of the spectrum. 5 PPTID was first defined by the WHO in 2007. 1 According to the mitotic activity and neurofilament staining, PPTIDs are classified as grade II (fewer than six cells undergoing mitoses with a positive neurofilament staining) or grade III (more than or equal to six cells undergoing mitoses with a negative neurofilament staining). 7
Takase et al. reported that microsurgery was the most suitable treatment option to achieve gross total resection. The decision to administer postoperative radiotherapy is based on the pathological grade, residual tumor, cerebrospinal fluid seeding, and clinical manifestations. Chemotherapy may increase the effect of radiotherapy, although its side effects remain uncertain. 3 In our patient, after the first surgery, there was no evidence of cerebrospinal fluid seeding. Although the tumor was believed to have been gross total resection during the first surgery, we still recommended radiotherapy at that time due to concerns about any small residual or possible further dissemination or recurrence. However, due to the patient’s reasons, she did not undergo radiotherapy at the time. According to Fauchon et al., the five-year survival rates for PPTID were 74% and 39% for grades II and III, respectively. Meanwhile, the recurrence rates were 26% and 56%, respectively. 8 The patient in the present case was postoperatively diagnosed with PPTID, WHO grade II. She had progression-free survival for 10 years, and the PPTID recurred 10 years after the first surgery.
Based on a literature review by Majovsky et al., there have been eight cases of pineal hemorrhagic apoplexy associated with pineal tumors. 4 The symptoms and signs of pineal apoplexy include headache, lethargy, unsteady gait, extraocular motor manifestations, and generalized hyperreflexia. 3 Some symptoms are exacerbated in the context of acute hemorrhage, resulting in hydrocephalus or midbrain compression. Pineal tumor apoplexy causes acute clinical deterioration of neurological function owing to acute intratumoral hemorrhage. 4 Various causes of pineal tumor bleeding have been identified. Apuzzo et al. reported that pineal tumor bleeding was likely related to anticoagulant therapy. 9 Burres et al. proposed other possible causes of pineal tumor hemorrhage, including hypertension, external or interstitial irradiation, craniocerebral trauma, tumor histology, tumor growth characteristics, hemorrhagic diathesis, and idiopathic causes. 10 Harada et al. reported a case of pineal tumor hemorrhage caused by an external ventricular drain or a ventriculoperitoneal shunt. 11 Matsumoto et al. reported a case of pineal tumor hemorrhage associated with ventriculoperitoneal shunt placement. 12 According to Wang et al., pineal tumor apoplexy may occur spontaneously. 13 Majovsky et al. reported that space-occupying lesions compress the surrounding structures of the pineal gland and interfere with the arterial blood supply and venous return of the pineal gland, resulting in bleeding. 4 The patient in the present case had no history of hypertension, trauma, or anticoagulant use. Although she underwent ventriculoperitoneal shunt insertion, the bleeding did not occur until 10 years later. The bleeding was mostly caused by the recurrent pineal tumor. Majovsky et al. compiled 30 cases of pineal apoplexy from the PubMed database, of which 8 are pineal tumors. 4 There were seven cases of pineal parenchymal tumors, and one of choriocarcinoma. 4
Cases of pineal tumor with apoplexy.

N/A, not applicable; HA, headache; HCP, hydrocephalus; SAH, subarachnoid hemorrhage; PPTID, pineal parenchymal tumor of intermediate differentiation.
Conclusions
PPTID is a rare disease that accounts for less than 0.1% of primary central nervous system tumors. Pineal apoplexy is rare, and its incidence and clinical significance remain unclear. We noted that pineal parenchymal tumors are more prone to tumor bleeding than other pathology types of pineal tumors. We proposed that this is attributed to the high vascularity of pineal glands and the pathological changes in pineal parenchymal tumors. There have only been eight reported cases of pineal tumors with hemorrhagic apoplexy. The recurrence of PPTID with apoplectic hemorrhage after 10 years has not been reported. Despite its rarity, PPTID with apoplexy should be considered in patients with PPTID presenting with sudden-onset neurological symptoms.
Footnotes
Acknowledgements
Author contributions
Yu-Li Chen, MD researched the literature and wrote the first draft of the manuscript. Li-Hsin Tai, MD researched the literature, collected information for series review and Table 1, and joined the article revision. Ann-Shung Lieu, MD, PhD participated in protocol development, obtaining ethical approval, patient recruitment, and data analysis. All authors reviewed and edited the manuscript and approved the final version of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
This case report was approved by the institutional review board at the Kaohsiung Medical University Hospital (Kaohsiung, Taiwan).
Informed consent
A signed patient permission form for medical research was obtained.