
Abstract
Background
Extraskeletal myxoid chondrosarcoma (EMC) is a rare malignant soft tissue sarcoma (STS) that accounts for less than 3% of all soft tissue tumors. The conventional treatment for primary EMC is wide local excision with or without radiation therapy.
Materials and Methods
This study was a retrospective review of all EMC cases treated within a single institution between 1992 and 2019. EMC was diagnosed using a combination of histologic morphology and immunostaining, with confirmatory fluorescent in situ hybridization. Overall survival (OS) and disease-specific survival (DSS) were defined using Kaplan–Meier analysis.
Results
Fifteen patients were evaluated, including 11 males and four females. The average age at presentation was 51.7 ± 20.4 years and the mean follow-up time was 61.5 months (range, 5–286 months). The average resected tumor size at largest dimension was 7.14 cm (range, 2.4–18.7). Twelve of fifteen (80%) patients underwent wide local excision, and nine of the twelve (75%) underwent local radiation therapy. The 1-, 5-, and 10-year OS was 80% (95% CI, 59.8–100), 72% (95% CI, 48.5–95.5), and 72% (95% CI, 48.5–95.5), respectively. The 1-, 5-, and 10-year DSS was 92.3% (95% CI, 77.8–100), 83.1% (95% CI, 61.5–100), and 83.1% (95% CI, 61.5–100), respectively. At last follow-up, 11 patients were alive and ten (90.9%) were disease free.
Conclusions
Extraskeletal myxoid chondrosarcoma is a very rare STS most often seen in males and in the extremities. Our cohort was too small to provide meaningful statistical analysis; however, we observed lower rates of local recurrence in patients treated with radiation.
Keywords
Introduction
Extraskeletal myxoid chondrosarcoma (EMC) is a rare, malignant soft tissue sarcoma (STS) with a predominantly myxoid histology that accounts for less than 3% of all soft tissue tumors. 1 EMC was originally believed to be a low-grade, slow growing subtype of chondrosarcoma. However, long-term follow-up studies have revealed high rates of local recurrence and distant metastasis more consistent with an intermediate- to high-grade tumor of unknown lineage.2-5 Similarly, improvements in molecular analysis have improved the ability to diagnose this rare entity. The male to female ratio is 2:1 and most patients are diagnosed between the ages of 40 and 59 years of age.3,5–7 EMC is commonly found in the lower extremities with a predilection for the proximal aspect of the extremities.2,4,8
Due to its rarity and unclear histologic origin, diagnosis of EMC depends more heavily on molecular genetics than many other tumors. EMC usually is characterized by gene abnormalities involving the Nuclear Receptor Subfamily 4 Group A (NR4A3) or the Ewing Sarcoma RNA Binding Protein 1 (EWSR1) genes and is most commonly characterized by a t(9;22)(q22;q12.2) chromosomal translocation. Approximately 70% of cases involve the NR4A3-EWSR1 fusion gene and it is believed that this fusion gene contributes to EMC tumorigenesis by affecting cellular growth and differentiation.1,6,7,9–11 The constitutive activation of NR4A3 is unique to EMC, thus making it a useful tool to differentiate EMC from histologically similar tumors such as myoepithelioma and myoepithelial carcinoma.1,6
The conventional treatment for primary EMC is wide local excision with or without radiation therapy, similar to most STSs.4,12,13 However, newer studies suggest a valuable role for radiation therapy in improving cancer-specific survival and reducing rates of local recurrence.2,3,7 Studies evaluating the role of chemotherapy in EMC have demonstrated poor response rates, thus relegating its use to a case-by-case approach.7,14
EMC has a higher rate of local recurrence and distant metastasis than other STS. Studies have demonstrated local recurrence rates ranging between 30 and 50% and distant metastasis upwards of 50%.4,7,8,12 Negative prognostic factors associated with local recurrence include larger tumor size, previous unplanned excision, a lack of radiation therapy, and high-grade tumors.1,2,6 Interestingly, increased recurrence rates do not correlate to worse overall survival (OS), which tends to be more favorable in EMC than other STSs.1,2 The reason for this is not entirely well understood.
Here, we review the current literature and describe our institution’s experience with EMC, including our treatment approach and outcomes for 15 patients. We demonstrate lower rates of local recurrence in patients treated with radiation.
Methods
Selection
Patient demographics and tumor characteristics.

EWSR1: Ewing Sarcoma RNA Binding Protein 1; FISH: fluorescence in situ hybridization; NR4A3: Nuclear Receptor Subfamily 4 Group A; SD: standard deviation.
Histopathological grading
Histological review of resected specimens was conducted by a musculoskeletal pathologist specializing in sarcoma at our institution. A primary diagnosis of EMC was made using a combination of morphology and immunostaining for EMA and cytokeratins. Confirmatory fluorescence in situ hybridization (FISH) was performed on thirteen of fifteen (86.6%) tumor samples. FISH assessed for gene break-apart signals in Nuclear Receptor Subfamily 4 Group A (NR4A3), performed by an outside institution, and Ewing Sarcoma RNA Binding Protein 1 (EWSR1), performed within our hospital.
Statistical analyses
Continuous and categorical data were analyzed using descriptive statistics. Overall survival and disease-specific survival (DSS) was defined using Kaplan–Meier methods and was calculated as the time from diagnosis or initial presentation at our institution to the most recent follow-up evaluation or date of death. Due to the small size of our cohort, we were unable to provide any statistically meaningful analysis.
Results
Patient demographic data
Thirty-two patients were initially identified via preliminary biopsy from 1992 to 2019 within our institution, but only fifteen met inclusion and exclusion criteria after histologic confirmation of EMC. Eleven of the patients were male and four were female. The average age at presentation was 51.7 ± 20.4 years and all patients presented with the primary complaint of a new mass (Figure 1). The mean follow-up time was 61.5 months (range, 5–286 months) 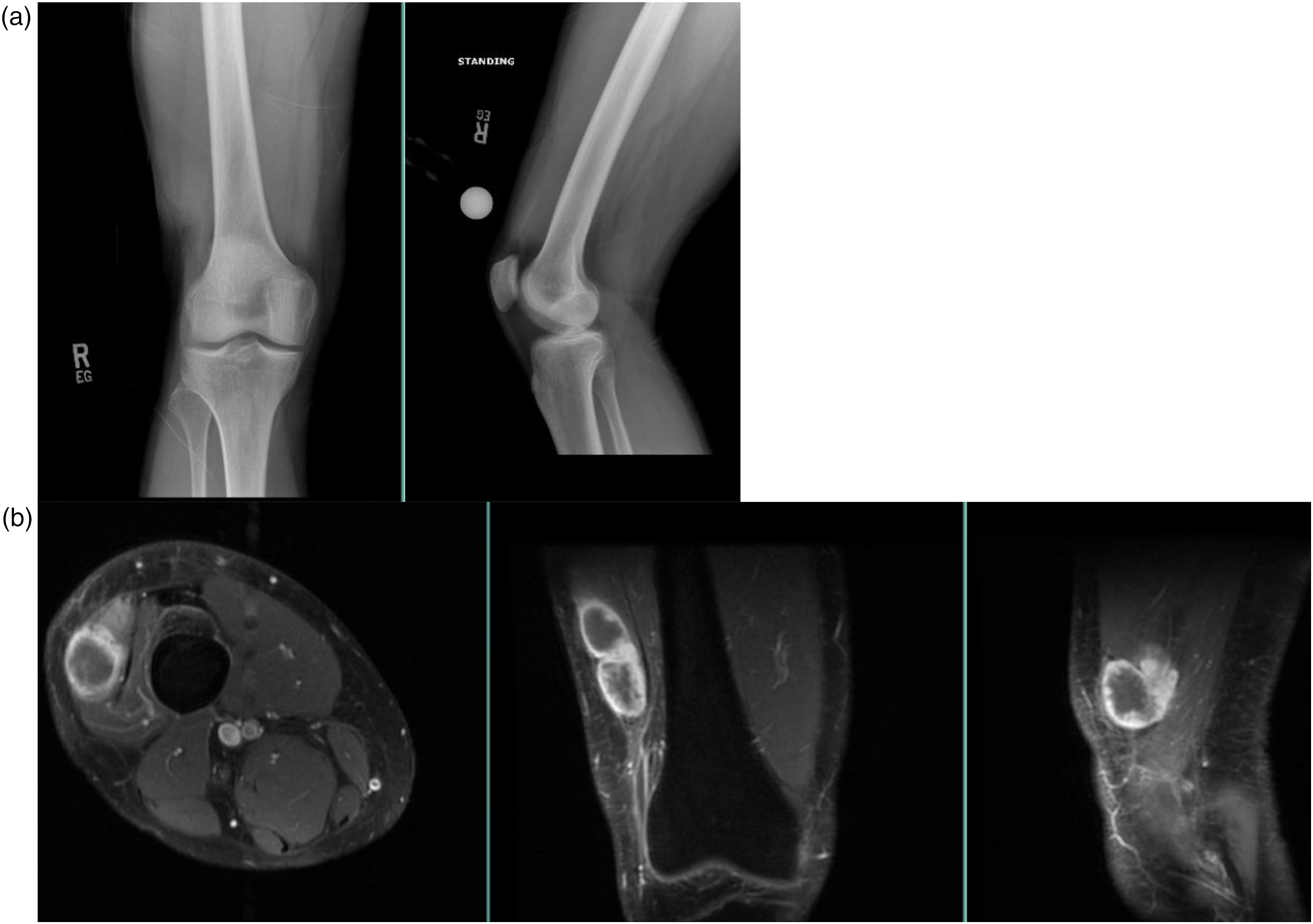
Histology and genetics
Extraskeletal myxoid chondrosarcoma was identified using a combination of morphology and immunostaining, with confirmatory FISH for the NR4A3 or EWSR1 gene break-apart signals (Figure 2). Fluorescence in situ hybridization (FISH) was performed on thirteen of fifteen (86.6%) tumor samples. FISH revealed gene rearrangement or genetic abnormalities in either one or both the NR4A3 or the EWSR1 genes in nine of thirteen (69.2%) patients Representative histology image for an extraskeletal myxoid chondrosarcoma, which is composed of interconnecting cords of tumor cells with eosinophilic cytoplasm with delicate elongated cytoplasmic processes. The tumor cells have uniform oval nuclei with evenly distributed chromatin. Abundant myxoid matrix is present in the background.
Location
Fourteen patients (93.3%) presented with disease localized to the extremities and one (6.7%) presented with axial disease localized to the posterior chest wall. Nine of fourteen extremity disease patients (64.3%) had lower extremity disease, and all were localized to the thigh and buttocks except for one patient with disease of the foot and ankle. The remaining five patients (35.7%) had upper extremity disease including one in the hand, two in the forearm, one in the arm, and one localized to the scapula (Table 1). The average resected tumor size at largest dimension was 7.14 cm (range, 2.4–18.7) (Table 1).
Treatment
Study characteristics.
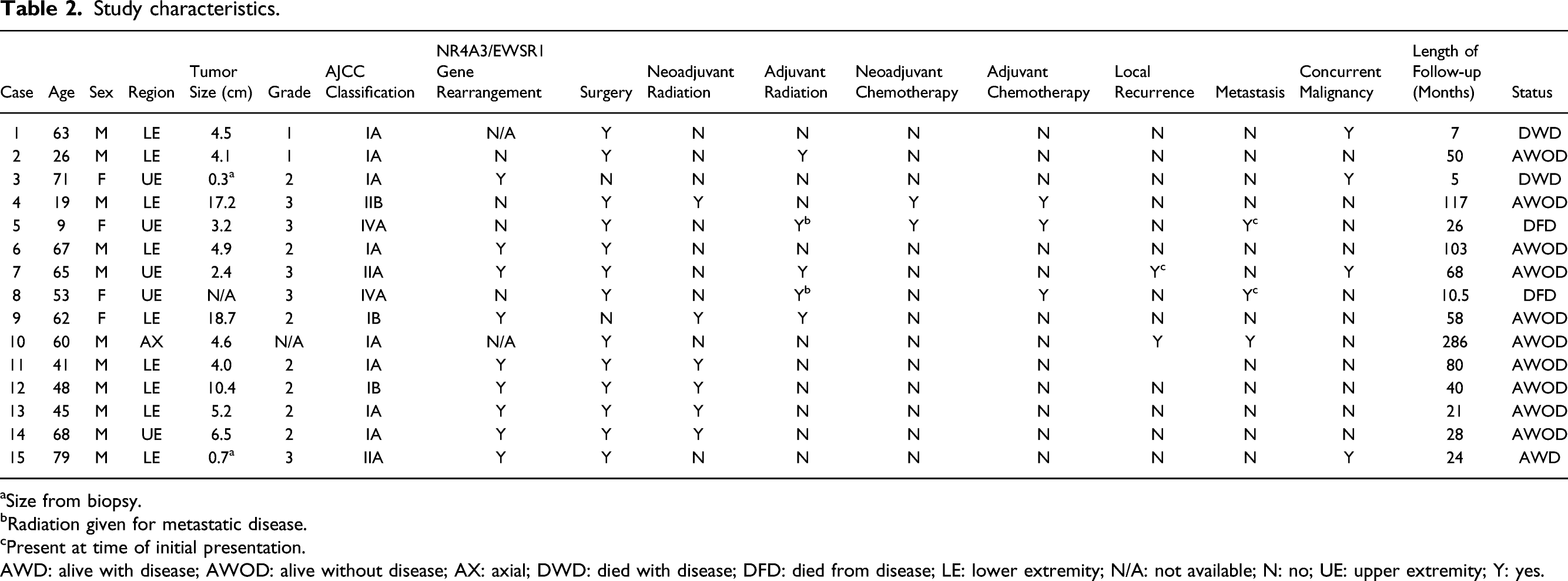
aSize from biopsy.
bRadiation given for metastatic disease.
cPresent at time of initial presentation.
AWD: alive with disease; AWOD: alive without disease; AX: axial; DWD: died with disease; DFD: died from disease; LE: lower extremity; N/A: not available; N: no; UE: upper extremity; Y: yes.
Neoadjuvant radiation was used in six of twelve (50%) patients, while three additional patients received adjuvant radiation therapy. Two patients that were less than 20 years old were treated according to our institution’s pediatric oncology recommendations and received neoadjuvant and adjuvant chemotherapy (16.6%). Additional treatment characteristics can be seen for each patient in Table 2.
Outcomes
The 1-, 5-, and 10-year OS in our cohort was 80% (95% CI, 59.8–100), 72% (95% CI, 48.5–95.5), and 72% (95% CI, 48.5–95.5), respectively (Figure 3(a)). Two patients with EMC died during follow-up from concurrent malignancies (Table 2). As a result, the 1-, 5-, and 10-year DSS was 92.3% (95% CI, 77.8–100), 83.1% (95% CI, 61.5–100), and 83.1% (95% CI, 61.5–100), respectively (Figure 3(b)).
Nine of twelve (75%) patients who underwent wide local excision also received neo- or adjuvant radiation therapy. Eight of nine (88.9%) patients who received radiation therapy remained disease free without local or distant disease recurrence. Only one patient (11.1%) experienced local recurrence or metastatic progression (Table 2). This involved four separate episodes of local recurrence, each achieving negative margin resection, and one instance of metastatic disease to the lungs requiring a metastasectomy. Time to original local recurrence was 11 years and the patient has been disease free for the past 4 years (Table 2).
At time of last follow-up, 11 patients were alive and ten (90.9%) were disease free. The only patient with active disease refused initial surgical excision and received local radiation therapy only. Both patients that presented with metastatic disease to the lungs died within 17 months (Table 2).
Discussion
EMC was originally described in 1972 by Enzinger and Shiraki as a low-grade sarcoma consisting of primitive chondroid cells. 15 EMC remains poorly understood, although long-term follow-up studies have demonstrated high rates of local recurrence and distant metastasis suggesting that EMC is better characterized as an intermediate- to high-grade tumor of unknown origin.2–5 Due to its rarity and unclear lineage, there are limited studies that have evaluated patient demographics and disease-related outcomes, including rates of local recurrence and distant metastasis.1–5,7,8,12–17 In our case series of 15 patients over 27 years, we showed that 88.9% of patients who underwent either neoadjuvant or adjuvant radiation therapy for primary local disease remained disease free at last follow-up, suggesting a possible role for radiation in reducing the rate of local recurrence in the treatment of EMC.
In our study, we identified 11 males and four females diagnosed with EMC at an average age of 51.7 years. These findings are consistent with the current literature which demonstrates an average age at time of presentation in the 5th and 6th decade and a 2:1 of males to females (Table 1).2,4,6,13
The t(9;22)(q22;q12.2) translocation has consistently been identified in EMC tumors and the presence of NR4A3 offers a unique diagnostic tool that helps to differentiate EMC from similar tumors. 18 In our study, we found gene rearrangement or genetic abnormalities in either one or both the NR4A3 or EWSR1 genes in nine of thirteen (69.2%) patients. However, this was only specific to NR4A3 in four of thirteen (30.7%) patients (Table 2). This rate is significantly lower than other studies, which have shown rates of NR4A3 gene abnormalities closer to 70–90%.1,6 Our lower rate may be related to poor specificity of the original probes used since all samples tested prior to 2010 were negative for NR4A3 gene rearrangement. An alternative explanation is that not all EMC tumors depend on the NR4A3 gene rearrangement. In fact, Hisaoka et al. found that aberrant co-expression of NOR1 and SIX3 is an alternative rearrangement seen in EMC tumors without NR4A3 gene rearrangement. 19 Unfortunately, these genes were not tested for in our cohort and we are unable to corroborate these findings due to the retrospective nature of this study.
The average tumor size in our study at largest dimension was 7.14 cm (range, 2.4–18.7), which resembles prior studies demonstrating mean tumor sizes ranging from 7 to 9 cm.1,9,13 The average size of STSs at time of presentation is 10.2 cm and studies have shown large size to be a poor prognostic factor with a 3.5-fold greater risk of mortality in tumors greater than 15 cm compared to those less than 5 cm.1,6,20 With regard to EMC specifically, Drilon et al. demonstrated that size greater than 10 cm is a poor prognostic indicator for OS; however, we did not appreciate this in our cohort. Three patients underwent resection of tumors greater than 10 cm (17.2 cm, 18.7 cm, and 10.4 cm) with either neoadjuvant or adjuvant therapy and all remained disease free at last follow-up (minimum of 5 years) (Table 2). The difference in our findings is possibly related to the use of neoadjuvant and adjuvant radiation therapy, which is becoming more of a standard of care for EMC. Alternatively, our length of follow-up may not have been long enough to appreciate recurrence since EMC has been shown to have a lengthy delay prior to recurrence.
Many studies have documented high rates of local recurrence and distant metastasis with EMC, with rates of local recurrence ranging between 30 and 50% and distant metastasis upwards of 50%.4,7,8,12 Initial theories for the high rates of recurrence were attributed to the historical low-grade classification given to EMC, which led to an early underutilization of adjuvant therapies. Furthermore, the indolent progression suggested it was more of a benign mass and many unplanned, incomplete excisions resulted in improper margins. 6 Twelve patients in our cohort presented with local disease only, while one presented with local recurrence and two presented with metastatic disease to the lungs. This trend resembles other studies demonstrating the lungs as the primary metastatic site for EMC.4,13 Contrary to previous studies, we appreciated only two patients with episodes of recurrence. One patient presented less than 50 days after incomplete excision at an outside hospital and underwent wide re-excision with free flap coverage and adjuvant radiation therapy. He has remained disease free for more than 5 years (Table 2).
The other patient developed four episodes of local recurrence and was treated with negative margin re-excision each time. During his last local recurrence, he was also found to have isolated metastatic disease to the lungs and underwent a metastasectomy and has remained disease free for the past 4 years. Interestingly, this patient had an initial disease-free period of 11 years. While this is an extensive period of disease-free survival, it is consistent with prior studies.8,12,17 Paioli et al. demonstrated median time to local recurrence of 66 months (range 16–125) and median time to distant relapse of 20 months (range 1–84 months). 1 These findings suggest a need for prolonged follow-up due to a tendency for EMC to recur late and questions whether patients treated for EMC should have unique follow-up guidelines compared to other STSs. Due to our low recurrence rate and small sample size, we are unable to comment on possible prognostic factors for recurrence.
Our 1-, 5-, and 10-year OS rates resemble other studies that have seen 5-year and 10-year survival rates ranging from 80 to 90% and 60 to 70%, respectively.2,4–7,12 However, our cohort demonstrated much better DSS and disease-free survival compared to other studies. One possible explanation for this is that other studies have shown much higher rates of patients presenting after unplanned, incomplete excision at an outside hospital. Only 13% of our patients presented after a “whoops” procedure compared to rates ranging from 30 to 40% in other studies.1,4
Nine of 12 patients underwent wide local excision with adjuvant radiation therapy, and 88.9% of those patients remained disease free without local or distant disease recurrence at last follow-up. Many recent studies have shown similar findings, including a trend toward better local control and OS in patients treated with wide local excision and radiation therapy compared to excision alone.3,4,7 These findings need further study but do suggest a valuable role for radiation therapy in the treatment of EMC. Radiation therapy may also have a role for palliative treatment to metastatic sites as shown by Ogura et al., who demonstrated prolonged, progression-free intervals of disease in their retrospective review of 23 patients. 16
This study has several limitations. Foremost, the retrospective design limits the conclusions that can be drawn from our data. In addition, we were unable to apply any meaningful statistical analysis to our findings due to the small size of the cohort. Our mean follow-up of 61.5 months resembles earlier studies, but we are limited by short follow-up with some patients. Three patients only have 2 years of follow-up due to recent diagnosis and two others died from concurrent malignancies within the first few months after diagnosis. Many studies have shown that EMC has a delayed progression to recurrence, which may explain the low local recurrence rate observed in our cohort. Furthermore, genetic testing was only obtained in 13 patients, and we had a low rate of NR4A3 positive samples which is contrary to other studies.
Conclusion
To conclude, our cohort of EMC patients is comparable to prior studies in patient age, sex, tumor location, and size. Unlike prior studies, however, we demonstrate a lower rate of local recurrence and metastasis, as well as an improved DSS. We also observed lower rates of local recurrence in patients treated with radiation. Due to the limitations of this study, we were unable to provide meaningful statistical analysis that might explain these results.
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: ATB: BMJ Case Reports: Editorial or governing board; Clinical Orthopaedics and Related Research: Editorial or governing board; exparel/pacira: Stock or stock Options; Journal of Oncology Practice: Editorial or governing board; Journal of Surgical Oncology: ad hoc reviewer; Lancet - Oncology: Editorial or governing board; Musculoskeletal Tumor Society: Board or committee member; Onkos Surgical: Paid consultant; Pediatric Blood and Cancer: Editorial or governing board; Rare Tumors: Editorial or governing board; Rush Orthopedic Journal: Editorial or governing board; Swim Across America Cancer Research Grant: Research support; SG: Onkos Surgical: Paid consultant; Stock or stock Options; USMI: Stock or stock Options. All other authors have no pertinent financial disclosures or pertinent conflicts of interest.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Author Contributions
MPF: data collection, analysis, drafting, editing, production, and revisions; LL: analysis, drafting, editing, and revisions; PK: data collection and analysis; AA: drafting and production; MRC: drafting and editing; CAG: editing, production, and revisions; SG: conception, oversight, and revisions; and ATB: conception, oversight, drafting, editing, and revisions.
Ethical approval and informed consent
Rush University Medical Center obtained individual Institutional Review Board approval with an approved waiver of consent prior to beginning any research efforts.
Availability of data
The data that support the findings of this study are available from the corresponding author upon reasonable request.
List of abbreviations
STS, soft tissue sarcoma; EMC, extraskeletal myxoid chondrosarcoma; OS, overall survival; DSS, disease-specific survival; NR4A3, Nuclear Receptor Subfamily 4 Group A; EWSR1, Ewing Sarcoma RNA Binding Protein 1; FISH, fluorescent in situ hybridization.