
Abstract
Extraskeletal myxoid chondrosarcoma is a rare soft tissue neoplasm that occurs predominantly in the soft tissues of the lower extremities. Herein we present a case of a 29 year old male who presented with bilateral femoral numbness believed to be the result of prior injury to his back. A magnetic resonance imaging revealed a mass in the T4-T5 epidural space compressing the spinal cord. Laminectomy was performed and the lesion removed piecemeal. The pathology specimen consisted of multiple fragments of dura involved by a myxoid neoplasm with a nodular growth pattern. The tumor cells were arranged in anastomosing cords and strands. Individual tumor cells were small, of uniform size and shape, with small hyperchromatic nuclei and scant eosinophilic cytoplasm. Immunohistochemical stains were performed which showed the tumor cells were diffusely positive for vimentin and focally positive for EMA, S-100 protein and cytokeratin, whereas they were negative for CD34 and CD99. Fluorescence
Case Report
Clinical summary
A 29 year old male presented with bilateral femoral numbness believed to be the result of a prior back injury. The patient was treated conservatively for 7 months at which point he suffered another minor injury to his back that resulted in exacerbation of his lower limb numbness. He was referred to an orthopedist for further evaluation of his symptoms. A MRI revealed a mildly lobulated, well-defined heterogeneous T2 hyperintense mass in the left epidural space causing compression and displacement of the spinal cord without evidence of bony destruction (Figures 1 and 2). A T4-T5 laminectomy piecemeal resection was performed for spinal cord decompression. According to the intraoperative report, the capsule of the mass was adherent to the lateral and anterior dura of the cord. An intraoperative frozen section was performed and a preliminary diagnosis of meningioma was made. A post-operative staging work up revealed no evidence of tumor elsewhere. Following surgery, the patient remained asymptomatic and reported that his pre-operative symptoms had abated. No adjuvant therapy was recommended. He remained asymptomatic 12 months after his initial surgery.

A) Axial T2-weighted imaging demonstrates a mildly lobulated, well-defined heterogeneous T2 hyperintense mass in the left epidural space causing compression and displacement of the spinal cord to the right. There is mild scalloping of the posterior aspect of the T4 vertebral body consistent with a long-standing slow-growing tumor. B) Axial pre- contrast T1-weighted imaging confirms a T1 hypointense intensely enhancing mass. There is no evidence for bony destruction or invasion.
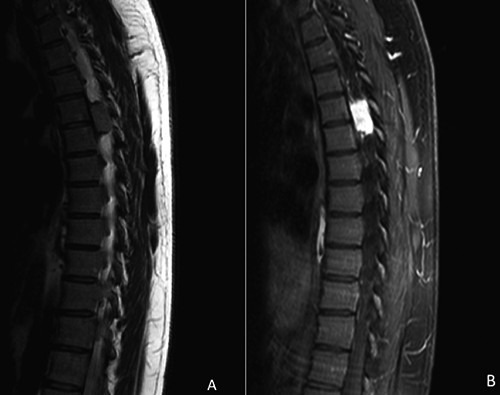
A) Sagittal T2-weighted imaging demonstrates a well-defined heterogeneous T2 hyperintense mass in the T4 and T5 left epidural space. B) Post- contrast T1-weighted imaging confirms a T1 hypointense intensely enhancing mass.
Pathologic findings
The laminectomy specimen consisted of multiple fragments of dura that were involved by a neoplasm with a nodular growth pattern (Figure 3A). The tumor cells were arranged in anastomosing cords and strands. Individual tumor cells were small, of uniform size and shape, with a small hyperchromatic nucleus and scant eosinophilic cytoplasm and were embedded in an abundant myxoid, basophilic matrix (Figure 3B). By immunohistochemistry, the tumor cells were diffusely positive for vimentin, focally positive for EMA, S-100 protein and cytokeratin (Figure 3C) and negative for CD34 and CD99. Fluorescence

A) Hematoxylin and eosin stained section (4x) demonstrating tumor cells embedded in a myxoid background with extensive infiltration of the dura. B) Hematoxylin and eosin stained section (4x) highlighting a nodular growth pattern. The individual tumor cells were small, uniform with small hyperchromatic nuclei and scant eosinophilic cytoplasm (Inset). C) Vimentin was diffusely and strongly positive in the tumor cells. D) Fluorescence
Discussion
Extraskeletal myxoid chondrosarcoma was first described by Stout and Verner,
1
who reported a group of tumors which they labeled as
Extraskeletal myxoid chondrosarcoma is a rare soft tissue sarcoma that comprises less than 3% of all soft tissue sarcomas. 4 These tumors have a male preponderance (M:F ratio 2:1) and generally occur in the 5th to 6th decades of life, although rare cases have been reported in children. They are most commonly located in the extremities, limb girdles or trunk. 5 Since its original description, EMC has been reported in unusual locations, including the nasal cavity, the central nervous system and the vulva amongst other sites. In the central nervous system, there have been fewer than 10 cases of EMC reported; predominantly in the intracranial location, with sites of origin including the choroid plexus, pineal gland and cortex.6–10 However, there have been only two prior reported case of what can be assumed to be an EMC arising in an intradural spinal location, although the diagnosis was not confirmed by molecular studies.11,12
The exact pathogenesis of EMC is unclear, however, there is recent evidence that suggests that these tumors arise from a primitive mesenchymal cell with a propensity for multidirectional differentiation.
13
The original hypothesis that this tumor represents a variant of chondrosarcoma has been challenged in the recent years. The absence of true cartilage formation, the presence of a myxoid matrix that is rich in mucin and poor in collagen II and aggrecan core protein, as well as the presence of neurosecretory granules, microtubule associated protein-2 and beta tubulin in some cases, have lent credibility to the notion that EMC has neural-neuroendocrine differentiation and is not strictly a cartilaginous tumor.
9
The fact that tumor cells are positive for vimentin in virtually all cases, but only positive for S-100 protein focally in a minority of cases, again points to a non-hyaline cartilage phenotype. Epithelial markers, GFAP, HMB-45, desmin and myoglobin are all typically negative. The overall prevalence of gene fusion in EMC is estimated to be approximately 75% as reported in the literature.
14
EMCs harbor a balanced translocation involving the
A recent study investigated the expression of SMARCB1/INI1 protein that is known to be involved in malignant rhabdoid tumor, another sarcomas with
Other myxoid lesions may enter into the differential diagnosis of EMC and need to be clearly separated due to unique therapeutic and prognostic implications. The low cellularity and abundant myxoid matrix associated with EMC may cause diagnostic confusion with certain benign lesions such as myxoma and nodular fasciitis. The presence of nuclear hyperchromasia and the typical septated, nodular growth pattern help in distinguishing EMC from the above-mentioned entities. In cases originating in the axial skeleton, as was the case in our patient, chordoma is the main differential diagnostic concern that must be separated from EMC. Chordoma can usually be distinguished from EMC by the presence of vacuolated physalipherous cells that are typically positive for S-100 protein, EMA, cytokeratin as well as brachyury. In contrast, EMC is typically negative for epithelial markers and expresses S-100 protein only focally, if at all. Recognition of variants of EMC, including predominantly cellular to solid tumors with minimal myxoid matrix (cellular variant), rhabdoid differentiation, or spindle cells resembling fibrosarcoma or pleomorphic sarcoma, is also extremely important. In cases composed predominantly of solid areas, the tumor may resemble other tumors with small round cells, including Ewing sarcoma and small cell synovial sarcoma. However, careful sampling of the tumor usually reveals areas with histologic features of conventional EMC. In cases where histologic distinction of EMC from the above-mentioned entities is difficult on morphologic grounds alone, studies to detect the presence of the characteristic gene fusion involving EWSR1, and the fusion partner in Ewing sarcoma, are invaluable in aiding in the accurate classification of this tumor.
Extraskeletal myxoid chondrosarcoma is typically associated with a protracted clinical course, even when metastases develop, and is hence thought to be best classified as a low grade sarcoma. In the largest series analyzing the prognostic parameters in soft tissue EMC, the authors studied the morphologic, clinical, immunohistochemical and ultrastructural features in 117 cases. 5 The estimated median survival time was 18 years and median intervals to metastases and local recurrences were 12 and 8 years, respectively. Metastases occurred in 46% of cases and most frequently were to the lungs, followed by soft tissues, lymph nodes and bones. The estimated 5-, 10-, and 15-year survival rates were 90%, 70%, and 60%, respectively. Although, there is limited prognostic information available about EMC arising in a spinal location, prior reports of EMC arising in an intracranial location have shown a high recurrence rate.10,12,20 En bloc resection is widely considered to be the optimum surgical approach with complete resection being associated with lower recurrence rates.21,22
Extraskeletal myxoid chondrosarcoma has a poor response rate to chemotherapy and does not display a consistent response to any of the known commonly used chemotherapeutic agents for soft tissue sarcomas. Aggressive local control of disease is the primary approach to management.
23
Recent investigation into the tumor biology of EMC has identified the
In conclusion, EMC is a rare soft tissue tumor that can occasionally occur as a primary dural based lesion in various locations within the central nervous system. Although rare, EMC should be considered in the differential diagnosis of myxoid lesions of the spinal cord. Molecular studies demonstrating involvement of the
Footnotes
Conflict of interests: the authors declare no potential conflict of interests.