
Abstract
Rare pediatric tumors are heterogeneous group containing a variety of histopathological diseases, they represent approximately 10% of all childhood cancers. These rare tumors had a diversity of histology and clinical behaviors that pose different challenges to the investigators. Exploring different pediatric rare tumors. The data were reviewed, retrospectively, through the medical records of seven rare pediatric diseases between 2012 and 2019. Giant cell fibroblastoma (GCF) presented as painless swelling in the trunk, positive for CD34 with PTEN gene mutation. Neuroglial heterotopic tissue presented in 7 days old girl with facial asymmetry and bulging in the oral cavity, maximal de-bulking was done, histopathology was positive for GFAP and S100p. Left side neck mass, surgically excised revealed non-metastatic salivary grand mucoepidermoid carcinoma. Follow up without any chemotherapy or radiotherapy for 5 years with complete remission. Mesenchymal chondrosarcoma (MCS) presented in maxillofacial bones by persistent nasal bleeding, HEY1-NCOA2 fusion gene confirmed the diagnosis. Extra-osseous Ewing sarcoma (EES) presented as rubbery painless swelling in the scalp with fusion transcript involving EWSR1-FL11. Juvenile xanthogranuloma (JXG) presented by butter fly like skin patch in the face with foamy histiocytes in upper dermis with few Touton giant cells, extensive systemic involvement of lung and bone marrow. Metastatic ovarian choriocarcinoma with choriocarcinoma syndrome received induction two different lines of chemotherapy and consolidated with autologous stem cell transplant. Seven pediatric rare tumors, with different aspects of challenges in diagnosis and management, despite the absence of formal protocols and rarity of other center experiences.
Keywords
Introduction
Pediatric cancer is considered as a rare disease. A total of 15,000 out of 1.6 million (<1%) of diagnosed cancer patients in USA were below 20 years old. 1 Pediatric cancer is a heterogeneous group of different histopathological diseases. The diversity of these histologic subtypes poses challenges to the physicians to clarify their biologic and clinical behavior. The definition of an infrequent or rare childhood cancer is complex and has been interpreted differently by various investigators. The European Cooperative Study Group for Pediatric Rare Tumors (EXPeRT) defines a rare childhood cancer as one that has an incidence rate ⩽2 per million per year, is not considered in clinical trials, or both. 2 Within the Children’s Oncology Group (COG), the Rare Tumor Committee has adopted a qualitative definition of a rare cancer based on some common features that include low prevalence in younger patients, higher incidence in adults, and epithelial (rather than mesenchymal) tumor origin. 3 According to International Classification of Childhood Cancer subgroup 11 of the SEER database, COG has defined rare childhood cancers as those classified as “other malignant epithelial neoplasms and melanomas”. 4 The histologic features of this subgroup include carcinomas, such as nasopharyngeal, adrenocortical, and thyroid carcinomas, as well as melanoma and other unspecified carcinomas. However, this definition does not include rare cancers seen almost exclusively in children (e.g. pancreatoblastoma and pleuropulmonary blastoma [PPB]). Defining a rare cancer based solely on epidemiologic or pathologic traits is problematic, and a collaborative effort to better refine this concept is desperately needed. 5 In this study, we aimed to draw attention for few of rare diseases, that may help the physicians to unify the guidelines of diagnosis and management.
Materials and methods
In our study, we retrospectively reviewed the medical record of the cancer registry from 2012 till 2019. Total of seven pediatric patients with rare tumors were diagnosed and treated at our hematology/oncology center, Prince Sultan Military Medical City (PSMMC), Riyadh, Saudi Arabia. Rare cases of leukemia and lymphoma were excluded. The medical records were reviewed for details of the clinical history, demographic and laboratory data, histopathological diagnosis, radiological findings, molecular testing, and clinical data including treatment protocol and outcome. This study was approved by the Institutional Review Board at PSMMC (HP-01-R-079).
Results
Case 1
A 32-month-old girl, known PTEN gene mutation, had macrophaly, delayed motor milestones, and congenital cardiac anomalies. She presented with painless swelling in the right axilla since birth but progressing in size over the last few weeks, with newly developed similar two swellings at the upper left and lower right back. The swellings were soft, not tender, freely mobile, and measuring 1.5 cm × 1.5 cm. Ultrasound showed well-defined cystic structure with no vascularity. Excisional biopsy was done, histopathological study confirmed diagnosis of giant cell fibroblastoma, positive CD34, with negative margins. Six months later, patient presented with new swelling at left side of abdominal wall, soft, not tender, mobile, and 2 cm × 2cm. Wide local excision of the swelling done, histopathology revealed the same diagnosis, giant cell fibroblastoma. The patient didn’t receive neither chemotherapy nor radiotherapy. The patient still under follow up with clinical examination every 3 months with no new lesions in the last 1 year.
Case 2
A 7-days-old baby girl, her parents noticed facial asymmetry with bulging appears in the right upper part of oral cavity. MRI head and neck showed heterogeneous mass lesion involving the right infratemporal fossa and extending to the para-pharyngeal space protruding within the oropharyngeal cavity, extension to the skull base at right middle fossa is noted with lytic process of the bone including the petrous and mastoid, however the whole extension remains extra meningeal with no evidence of intracranial extension (Figure 1(a) and (b)). Bone scan and whole-body CT revealed no metastasis. Incomplete excision of the mass was done from multiple areas of the mass and histopathology was consistent with neuroglial heterotopic tissue, positive for GFAP and S100p. The patient didn’t receive any treatment and started follow up by MRI every 6 months. After 3 years of follow up, last MRI revealed reduction in the size of cystic components of the lesion seen in the complex mass slightly pushing the right tonsil toward the midline.
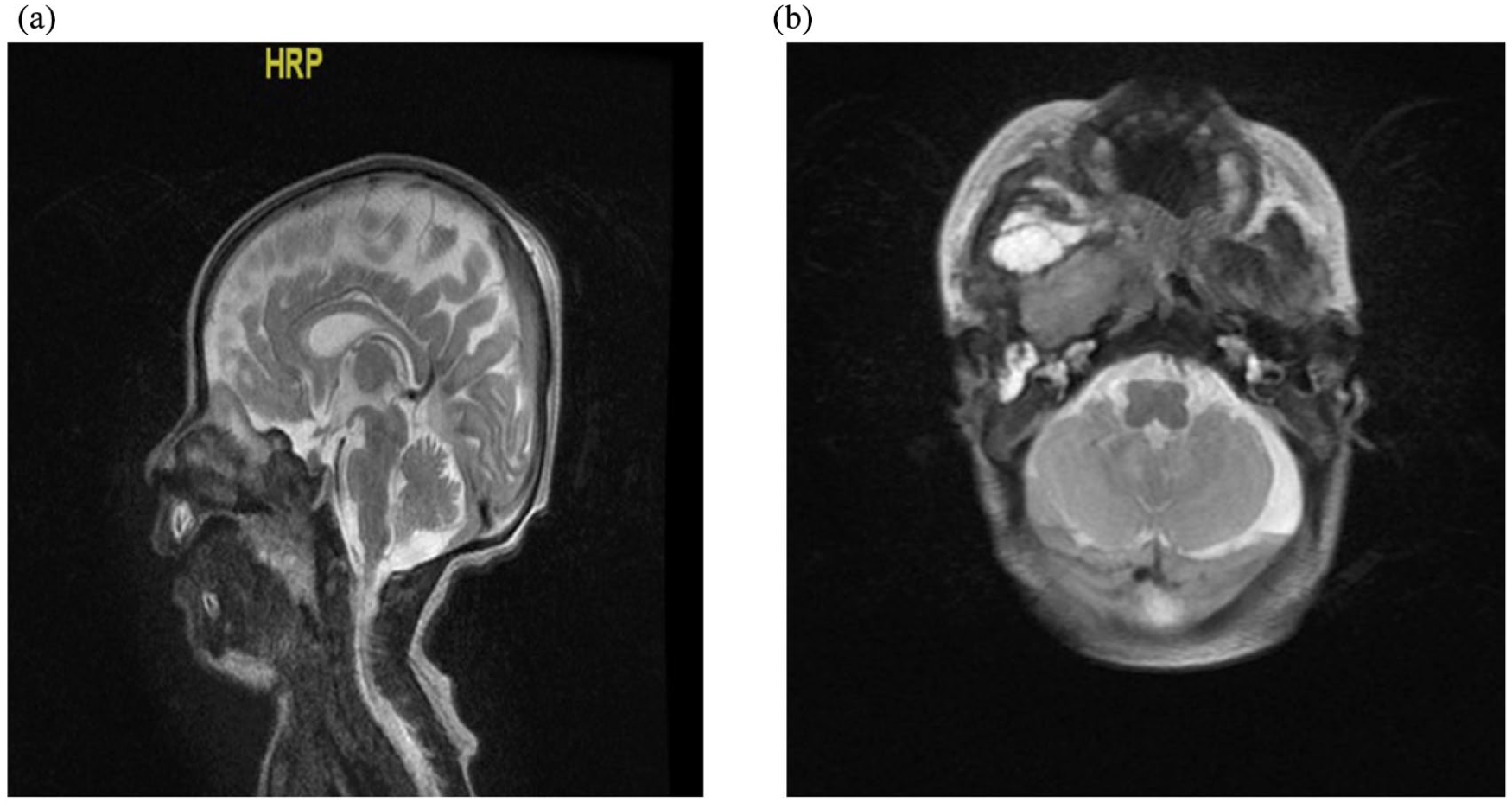
(a) and (b) Magnetic resonant imaging on head and neck showing right infra temporal and parapharyngeal region represent multiloculated cystic mass with postero-superior thick keratineous/proteineceous content.
Case 3
A 3-years old female patient presented with swelling in the left side of the face. It was slowly growing painless mass, located over the parotid gland, not attached to overlying skin. CT neck and maxillofacial showed salivary gland mass, no cervical LNDs enlargement. The mass was surgically removed and histopathology revealed mucoepidermoid carcinoma in salivary gland, low-grade, mitotic index is <4/10 high power field (HPF), inked surgical margins are involved by the tumor. Negative for vascular or perineural invasion in the tissue examined. Rim of normal salivary gland tissue noted. Post-operative local MRI revealed no evidence of abnormal soft tissue mass in the left neck region. Post-operative PET CT done showed absence of significant hypermetabolic activity at the site of known excised mucoepidermoid carcinoma in the left neck. Although the surgical margins are not free but our recommendation was to put the patient under follow up, without chemotherapy or radiotherapy, because it is low grade tumor with low mitotic index. Follow up, for the next 4 years, by ultrasound neck every 3 months, revealed no evidence of local recurrence.
Case 4
A 12-year-old female patient presented with unexplained epistaxis for 2 months. Blood tests showed normal complete blood picture (CBC) and coagulation profile. Intranasal endoscopic examination revealed a non-tender soft tissue outgrowth from right turbinates. Maxillofacial MRI showed a mass in the right nasal cavity involving all right turbinates (Figure 2(a)). Other body scanning by CT and bone scan were negative for any metastasis. Bilateral bone marrow biopsies were negative for malignancy. The patient underwent gross total resection of the mass. Histopathological examination exhibits a biphasic appearance, sheets of undifferentiated small blue round and spindled mesenchymal cells, interspersed with islands of hyaline cartilage (Figure 2(b)). The undifferentiated mesenchymal cells have an oval to elongated hyperchromatic nuclei and scanty cytoplasm. A prominent hemangiopericytoma—like vascular pattern was evident. Foci of osteoid matrix were also present. Immunohistochemical studies showed positive bcl-2, weakly positive CD99, and focally positive Desmin, Myo-D1, smooth muscle actin, and CD56. Other markers including cytokeratins, epithelial membrane antigen, CD34, CD45, CD20, synaptophysin, chromogranin, calponin, OCT3/4, and NSE markers were negative. Co-expression of Desmin and myod1 are indicative of rhabdomyoblastic differentiation. RT-PCR testing for HEY1-NCOA2 gene fusion was positive confirming the diagnosis mesenchymal chondrosarcoma. Second look surgery was done to ensure negative margin. The patient didn’t receive any adjuvant chemotherapy nor radiotherapy and she started follow up with MRI every 3–6 months. She is under follow up for 3 years without any local recurrence.
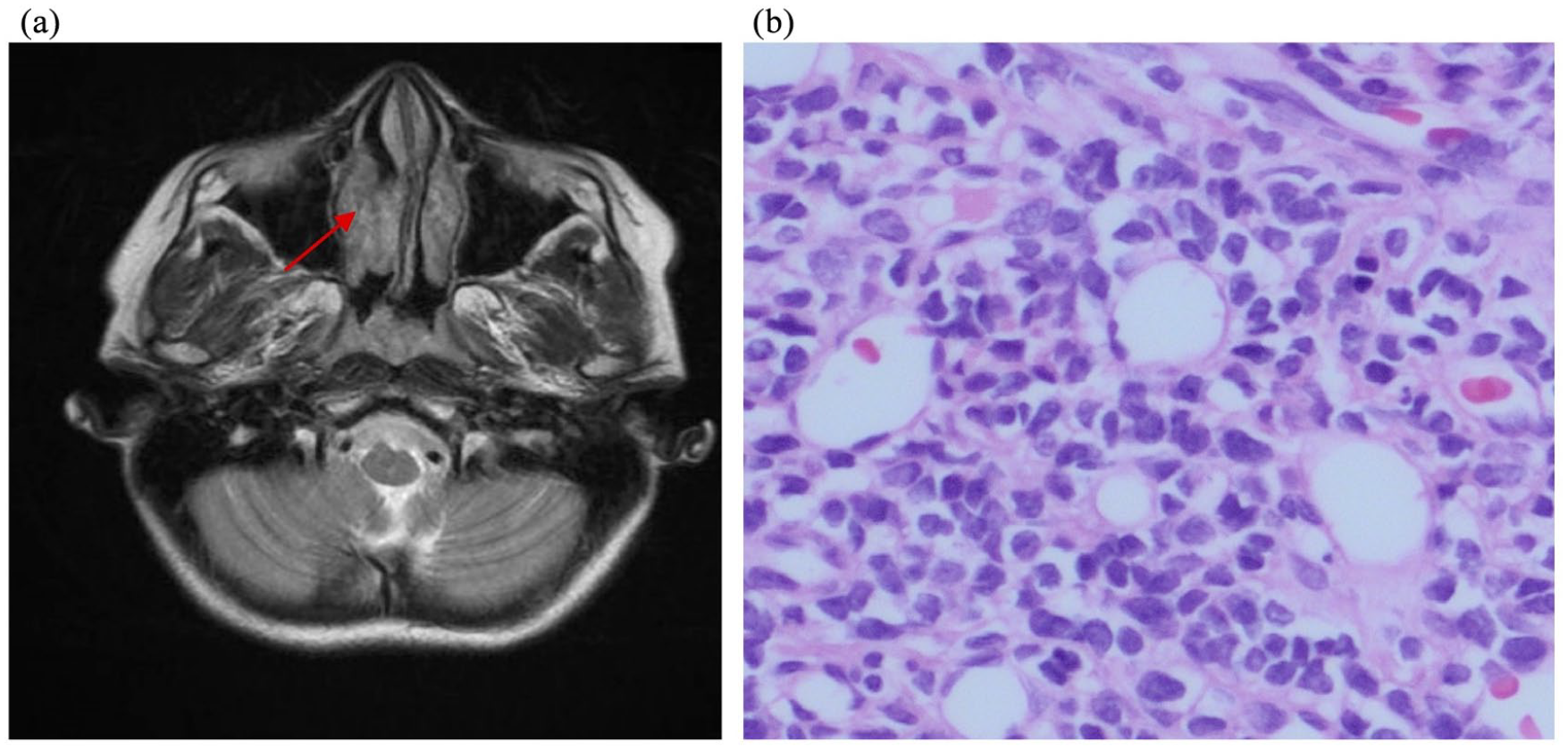
(a) Initial imaging MRI showing a mass in the right nasal cavity involving right turbinate and (b) Photo micrograph showing the biphasic component of mesenchymal chondrosarcoma. In the upper half of the image there is the undifferentiated small blue cell component, and in the lower half a hyaline cartilage is seen (hematoxylin-eosin, original magnification ×200).
Case 5
A 16-month-old girl, presented with painless progressing swelling in the scalp, it was rubbery, not tender, measuring 4 cm × 5 cm. Systemic examination was unremarkable. Head magnetic resonance imaging (MRI) (Figure 3(a)) showed complex lesion mainly within the scalp overlying the right frontal bone with solid and cystic component with no bony defect. Other metastatic work up in the form of computed topography (CT) scanning, bone marrow biopsy bone and PET scanning were negative. Complete surgical resection done (Figure 3(b)). Histopathological study showed subcutaneous cystic structure lined by small round blue cells with low grade morphology, positive for Vimentin, CD99, CD31 (focal), and PAS (focal) and negative for EMA, CK, CAM5.2, Actin, Desmin, Myogenin, Myo-D1, Synaptophysin, S100, CD34, and CD45. Surgical Margins were negative less than 1 cm. Cytogenetic analysis showed fusion transcript involving EWSR1-FL11. The patient was diagnosed as “Non-metastatic Extra-osseous Ewing sarcoma (EES)”. ES protocol “EURO-E.W.I.N.G. 99” was started. After six cycles of chemotherapy, time of local control, second look surgery was done to ensure wide negative margins. Radiotherapy was not received. Chemotherapy protocol was completed. Follow up with local MRI every 3–6 months, for more than 5 years, showed no local recurrence.

(a) MRI head and neck showing a complex lesion mainly within the scalp overlying the right frontal bone which on the T2 weighted sequence and (b) post-operative MRI head &neck showing complete resection of the tumor.
Case 6
A 4-years-old male, presented during the neonatal period by non-tender butter fly like skin patch in the face, yellow brown in color with thick consistency. Topical steroid didn’t improvement it. At the age of 2 years, he was referred to oncology department because of prolonged fever, pancytopenia, and hepatosplenomegaly. Skin Biopsy from right cheek showed foamy histiocytes in upper dermis with few Touton giant cells (Figure 4(a)), positive for CD68 (Figure 4(b)), and negative for CD1a, S100, and Langerin. Bone marrow aspirate and biopsy showed histiocytic proliferation with similar immunophenotyping profile (Figure 4(c)). The patient was diagnosed as “xanthogranulomatosis”. CT lung showed bilateral basal ground glass appearance (Figure 4(d)). Abdominal ultrasound showed hepatosplenomegaly. Brain MRI showed bilateral mucosal thickening of maxillary and ethmoidal sinuses but no hypophyseal affection. Normal ophthalmology and endocrinology examinations. The patient started Langerhans cell histiocytosis III (LCH-III) protocol. Evaluation after 6 weeks induction showed progressive disease with more lung infiltrate, prominent mesenteric lymph node, and persistent bone marrow xanthogranulomatosis. The chemotherapy was intensified to high dose cytarabine. After four cycles, the disease showed good clinical response with improvement radiological imaging of lungs, abdomen, and normal bone marrow. Few months later, the imaging showed progressive disease. The patient started third line chemotherapy cladribine/cytarabine (Ara-C) for two cycles. He went into complete remission which maintained for 1 year of follow up.
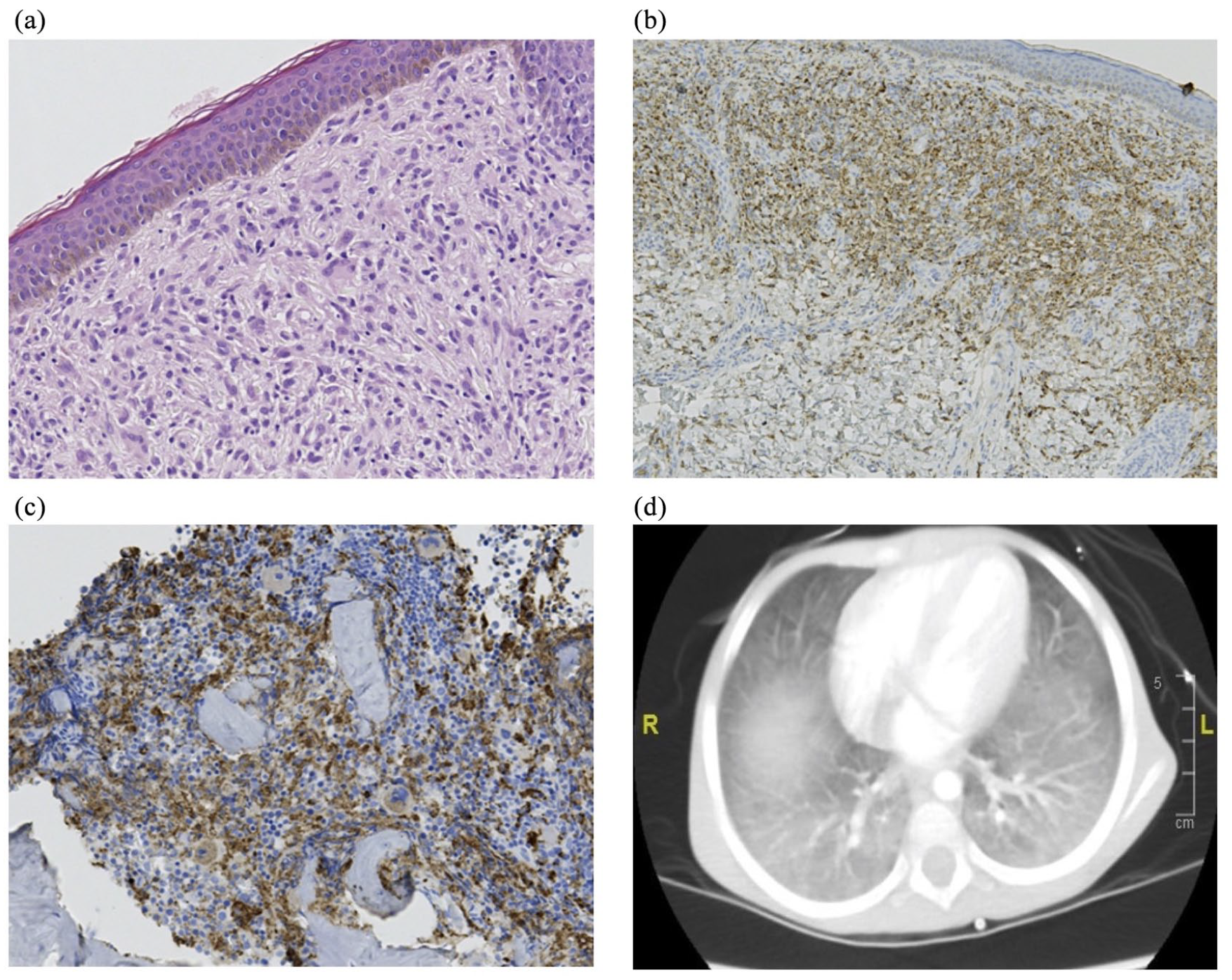
(a) Skin biopsy from right cheek showed foamy histiocytes in upper dermis with few Touton giant cells, (b) the histocytes express CD68-PGM1, (c) The BM biopsy show histiocytic proliferation highlighted by CD68-PGM1, and (d) CT lungs showed bilateral lung infiltrate.
Case 7
A 13-years-old girl presented with intermittent abdominal pain. Examination revealed diffusely mild tender abdomen with no definite masses. CT scan showed multiple cannon balls in both lungs 3.5 cm (Figure 5(a)), 1 cm hypodense lesion of the lower pole of the left kidney (Figure 5(b)), gastric mass in the fundus of the stomach, and huge mass raising from right ovary (Figure 5(c) and (d)). CT brain and bone scan were normal. Serum β-HCG was significantly high (226,066 IU/L), while AFP was normal (<0.5 kIU/L). Maximum de-bulking was done with residual pelvic mass. Few days later, the patient developed fresh hematemesis and hemorrhagic shock controlled by urgent GIT endoscopy. Couple of days later, the histopathology revealed Non-gestational ovarian choriocarcinoma (NGOC), Immunohistochemistry stains showed positive β-HCG, HPL, PLA, and pan-CK and negative CD117, OCT4, and CD30. The patient received two cycles of “PEB” (Cisplatin 20 mg/m2/dose × 5 days, Etoposide 100 mg/m2/dose × 5 days, Bleomycin 15 units/m2/dose × 1 day). Evaluation showed dramatic decrease of β-HCG 290 IU/L but the imaging scan showed mixed response. The chemotherapy was intensified to C-PEB with adding of cyclophosphamide 1200 mg/m2/dose × 1 day. β-HCG, after four cycles of C-PEB, became normal (<1.0 IU/L). CT evaluation showed regression of the primary pelvic lesion, renal lesions, gastric lesion. and chest nodules. But there were new three basal lung nodules which were not seen before. Second line regimen VeIP was recommended (vinblastin 0.11 mg/kg/day × 2 days, Ifosfamide 1200 mg/m2/day × 5 days, Cisplatin 20 mg/m2/day × 5 days). Evaluation after two cycles revealed further regression in the pelvic mass and pulmonary metastases but unchanged renal metastatic lesions. Another two cycles, total four cycles VeIP. Evaluation showed stable small focal pelvic lesion, resolved pulmonary metastasis, and kidney lesion. This good response was consolidated by high dose chemotherapy, carboplatin 700 mg/m2 and Etoposide 750 mg/m2 on day-5, day-4, and day-3, followed by autologous stem cell rescue. She discharged at day +17 post stem cell rescue with perfect general condition. Now, she is 4 years follow up with β-HCG and images with complete remission.

(a) CT chest showed multiple cannon balls (max 3.5 cm), (b) CT abdomen showed 1 cm hypodense lesion of the lower pole of the left kidney, and (c) and (d) CT pelvis showed huge mass raising from right ovary.
Discussion
Case 1
Giant cell fibroblastoma (GCF) is a unique fibroistiocytic childhood tumor of intermediate grade, commonly affects the trunk, more predominant in males younger than 10 years. GCS has high local recurrence (50%), with no reported distant metastasis. 6 Germline mutations of PTEN are the underlying genetic causes of related disorders clinically referred to as PTEN hamartoma syndromes (PHTS). These Mutations result in a non-functional or absent protein, which causes uncontrolled cell growth, leading to tumor (either benign or malignant) growth. 7 Giant cell Fibroblastoma and Dematofibrosarcoma Protuberans (DFSP) are closely related. Morphologic features of both GCF and DFSP may coexist within the same lesion, called hybrid lesions. 8 Both tumors are consistently CD34 positive and both share the t(17;22) (q22;q13) translocation resulting in the COL1A1-PDGFB gene fusion. 9 Treatment is wide local excision however, there is no standardization of the adequate surgical margins, micrographically oriented histographic surgery (MOHS) had also been tried. 10 One study concluded that Inhibition of in vitro growth of GCF cells using platelet-derived growth factor (PDGF) receptor antagonist, suggesting a potential treatment with the PDGF receptor tyrosine kinase inhibitor. 11 The European Organization for Research and Treatment of Cancer (EORTC) is leading a current clinical trial for assessment of imatinib mesylate in treating patients with locally advanced or metastatic dermatofibrosarcoma protuberans or giant cell fibroblastoma (NCT00085475).
Case 2
Neuroglial heterotopia has been defined as a mass composed of mature brain tissue isolated from the cranial cavity or spinal canal. 12 This anomaly can occur in the nasal cavity, scalp, orbit, pterygopalatine fossa, pharynx, palate, lips, tongue, middle ear region, and even lung. 13 Nasal heterotopic brain tissue masses often present with upper airways obstruction and, mostly when located in the para-pharyngeal space and when obstructing the oral cavities, it manifests with stridor and respiratory distress or feeding difficulties. 14 In a minority of cases, heterotopic glioneuronal tissue presents as a cystic lesion, likely due to the presence of functioning choroid plexus epithelium, these cystic lesions can expand rapidly and are more likely to cause airway obstruction or acute respiratory distress. 15 MRI is useful not only in identifying the heterotopic neural tissue extension, but also in excluding associated cranial defects and direct communication with the brain; thereby, they can be helpful for choosing the less hazardous surgical plane. 13 The distinction between the nasal glial heterotropia and encephalocele is impossible by pathologic means, as both lesions comprise the same types of neuroglial tissue. 16 Surgical resection is necessary in patients with nasopharyngeal lesions. It should be performed as early as possible in the newborn period to allow for extubation and oral feeding. However, patients should be reassessed with long-term follow up imaging, as these masses have the potential to increase in size or recur. 15
Case 3
In spite of mucoepidermoid carcinoma (MEC) is the most common malignant salivary neoplasm in adults, its incidence in the pediatric age group is extremely rarely. 17 Nearly 60% of MECs occur in the major salivary glands and 35% in the minor glands. 18 Histologically, MEC is sub classified to high and low grade. High-grade type is very aggressive tumor with high incidence of early metastasis and local recurrence.19,20 It is consisting of mucus-producing, squamous, and intermediate type cells as it implies, about two-third arise in the parotid gland, and one-third arise in the minor salivary glands. 21 The best therapy for of MEC consists of surgery with wide local excision, in some instance of difficulties in surgery, Platinum-based chemotherapy may be used. 22 However, routine radiotherapy is not recommended for the treatment of MECs. 23 The prognosis is depending mainly on tumor-free margins and the histopathological grading. 23
Case 4
Mesenchymal chondrosarcoma (MCS) is an extremely rare tumor with tendency toward late relapses and poor survival outcomes as low as 50% in 10-year-survival. 24 MCS is rarer than classic chondrosarcoma (CCS) and usually affects younger patients, more aggressive with local recurrence, while extra-osseous affection occurs in less than 1% in CCS.25,26 MCS is characterized by a biphasic histologic pattern of small undifferentiated round cells intermixed with islands of well-differentiated cartilaginous matrix. 24 Radical resection, with negative margins, of MCS is the cornerstone of management. Role of chemotherapy and radiotherapy is yet not well established. 27 Dantonello et al reported series of 15 MCS patients, the median age was 16.6 years and median follow-up 9.6 years. Four MCS were osseous and 11 extraosseous. All tumors were resected, but only eight completely resected without any residuals. About 13 individuals received chemotherapy, six were irradiated. 24 Ten-year event-free and overall survival rates were 53% and 67%, respectively. Four recurrences occurred, all within 4 years from diagnosis. One of these patients survived after surgery and radiation for local recurrence. Seven of eight patients whose tumors were completely resected during primary treatment, but only four of seven patients with incomplete surgery survived disease-free. 24 Less information is available about the most appropriate treatment for patients with metastases at presentation and further studies are required. 28
Case 5
Extraosseous Ewing sarcoma (EES) is an important subtype of Ewing sarcoma. 29 EES has Bimodal age incidence, older than 35 years and less than 5 years. 30 Few studies reported that its outcome is near to Ewing sarcoma of bone, if appropriate therapy was given. 29 Other studies reported that, there are differences between EES and skeletal ES in clinical behavior that may suggest underlying biological or genetic differences.29,31 Data about treatment and prognostic factors are limited to few studies that compare between osseous and extraosseous Ewing sarcomas.29,32,33 The prognosis of localized EES is poor prior to 2 years from initial diagnosis, but after 2 years the outcome is significantly better. 29 Although our patient was 16-month-old at initial diagnosis, but she had no local recurrence for the 5 years of close follow up.
Case 6
Juvenile xanthogranuloma (JXG) is a pediatric histiocytic disorder, it occurs predominantly in infancy or early childhood. 34 JXG characterized by xanthomatous skin lesions that may resolve spontaneously, systemic involvement may be life threatening. 35 JXG was under diagnosed due to its benign course and rarity. 36 JXG has been classified as the most common form a non-langerhans cell histocytosis. 37 In 85% of cases, it is possible to observe Touton giant cells, resulting from the fusion of macrophages and characterized by a crown of nuclei, with homogeneous eosinophilic cytoplasmic center, and prominent peripheral xanthomatization. 36 It is very rare that systemic JXG involves the bone marrow. As to our knowledge only nine cases of JXG with bone marrow involvement were reported.38,39 The prognosis of patients with exclusively cutaneous involvement is excellent, with spontaneous regression in months or even years, and relapses are rare. Cases with extra-cutaneous involvement, therapeutic approach is not consensual and should be evaluated on a case-by-case. Therapeutic options include chemotherapy, radiotherapy, and surgical excision, which should be considered casuistically. 36
Case 7
Non-gestational ovarian choriocarcinoma (NGOC) is an exceedingly rare germ cell neoplasm. It composes less than 0.1% primary ovarian neoplasms in a pure form. 40 Although the extreme rarity hinders an exact therapeutic outcome, a few cases with advanced NGOC with distant metastasis have occasional survivals. 41 NGOC has a poorer prognosis than gestational cases. Rarely, it can manifest by choriocarcinoma syndrome which is hemorrhagic manifestations of metastases. 42 Most reports have shown that choriocarcinoma syndrome typically occurs on day 2 or 3 after the beginning of chemotherapy from any site of metastasis, However, in our patient it occurred before chemotherapy in a form of hemorrhage from gastric mass. 43 Choriocarcinoma syndrome needs early recognition and urgent multimodal treatment. 44 To prevent or control this fatal syndrome, BEP regimes have been reported to be effective. Massard et al. 45 reported that the mortality decreased from 10/15(66.7%) to 2/10(20.0%) patients after BEP, but no data available about long term follow up and incidence of recurrence. In another study by Goswami et al., 46 2-year survival rate was 81% in patients who underwent surgery combined with chemotherapy, and 28% in patients who underwent surgery alone. In our case, we used PEB and C-PEB regimens to induce remission and to control choriocarcinoma syndrome, it was effective but due to delayed response, we consolidated this remission by autologous stem cell transplant. She is still in complete remission after 4 years of follow up.
Conclusion
Giant cell fibroblastoma is considered the juvenile form of DFSP, as both have the same translocation, wide local excision with a safety margin is the current treatment of choice. Heterotropic glial tissue mass should be included in the differential diagnoses of para-pharyngeal masses in the neonatal period. Mucoepidermoid carcinoma (MEC) should be considered in the case of a painless, slow-growing, parotid swellings. Mesenchymal chondrosarcoma (MCS) can be originate in unexpected sites and needs a high index of suspicion to diagnose. HEY1-NCOA2 fusion gene confirm the diagnosis. Extraskeletal Ewing’s sarcoma (ESE) should be considered in the differential diagnosis of subcutaneous masses in children and adolescents in spite of being rare. Localized xanthogranuloma (JXG) is rarely progressed to involve bone morrow. More studies needed to clarify the association between JXG and involvement of bone marrow. Choriocarcinoma syndrome is a fatal complication of advanced NGOC. High dose chemotherapy followed by autologous stem cell transplantation may consolidate the remission for a longer period.
Footnotes
Acknowledgements
The authors would like to thank SANAD Children’s Cancer Support Association for helping and support the research of children with cancer.
Author contributions
All authors have contributed to the manuscript in significant ways, have reviewed and agreed upon the manuscript content. Yasser Elborai: study concepts and design, manuscript preparation, manuscript editing. Nawaf Alkhayat: conceptualization, manuscript preparation, manuscript editing. Ghaleb Elyamany: conceptualization, guarantor of integrity of the entire study, experimental studies / data analysis, statistical analysis. Mohammad Alshahrani: literature research, supervision, manuscript preparation. Walid Ibrahim: literature research, supervision, manuscript preparation. Mohamed Othman: literature research, supervision, manuscript preparation. Hasna Hamzi: literature research, clinical studies, experimental studies / data analysis, supervision. Amal Binhassan: literature research, clinical studies, experimental studies / data analysis, supervision. Mansour S Aljabry: manuscript preparation, manuscript editing, supervision. Raniah Alqawahmed: manuscript preparation, manuscript editing, supervision. Yasir alrusayni: literature research, manuscript preparation, manuscript editing. Khadijah Abdulhaleem: literature research, manuscript preparation, manuscript editing. Omar Alsuhaibani: literature research, manuscript preparation, manuscript editing. Omar Alsharif: clinical studies, literature research, manuscript editing.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Ethical approval for this study was obtained from the Institutional Review Board at Prince Sultan Military Medical City (PSMMC)(approval number: HP-01-R-079).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by SANAD Children’s Cancer Support Association (grant number: RGP-2017-12)
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.