
Abstract
Myxoinflammatory fibroblastic sarcoma is a rare malignant soft tissue neoplasm that typically arises on the distal extremities of adults. It usually behaves in a low-grade manner and its characteristic histology is of a lobulated proliferation of moderately atypical spindled to epithelioid cells, vacuolated cells, and enlarged or bizarre cells with prominent nucleoli, dispersed within myxoid stroma containing a mixed inflammatory cell infiltrate. The etiology of myxoinflammatory fibroblastic sarcoma remains unknown with no definite causal factors identified. We describe a case of myxoinflammatory fibroblastic sarcoma arising in the foot of a 77-year-old female, which rapidly recurred locally after initial excision and which arose 10 years after renal transplantation. The neoplasm also showed intermingled areas of hemosiderotic fibrolipomatous tumor. The patient also had multifocal areas of squamous cell carcinoma in situ of the foot and hand, in keeping with the clinical context of immune deficiency. This is the second case of myxoinflammatory fibroblastic sarcoma reported to occur after transplantation, but additionally shows hybrid features of hemosiderotic fibrolipomatous tumor, highlights immunocompromise/immunosuppressive therapy as a possible etiologic factor in their development, and adds to the growing number of myxoinflammatory fibroblastic sarcoma that has demonstrated aggressive behavior.
Keywords
Introduction
Myxoinflammatory fibroblastic sarcoma (MIFS) is a rare malignant soft tissue tumor that most frequently arises on the distal extremities of young to middle-aged adults. MIFS typically behaves as a low-grade sarcoma with a high local recurrence rate, but very low metastatic rate1,2 with only small numbers of regional nodal and distant metastases reported.3–8 Histologically, MIFS is characteristically a multinodular lesion composed of three cell types: variably atypical spindle to epithelioid cells, vacuolated fibroblasts, and large pleomorphic virocyte-like or Reed–Sternberg-like cells1–3 within prominent myxoid stroma containing a mixed inflammatory cell population and variable hemosiderin deposition and fibrosis. The etiology of MIFS remains unknown, although some are associated with characteristic genetic abnormalities. We describe a case of MIFS occurring in the foot of a 77-year-old female, which also showed hybrid features of hemosiderotic fibrolipomatous tumor (HFLT), demonstrated rapid local recurrence, and was associated with a history of renal transplantation, highlighting a possible association of MIFS with a background of immunocompromise.
Case history
A 77-year-old female presented with a 2 month history of a mass on the dorsal aspect of the right foot. Her past medical history included atrial fibrillation, bilateral hip replacements, and renal transplantation for polycystic kidney disease 10 years previously, for which she took sirolimus (1 mg alternate days) and prednisolone (5 mg daily). On clinical examination, the right foot mass was presumed to be a hematoma. She underwent evacuation of the hematoma at her local institution, but the histology unexpectedly showed a spindle cell neoplasm with focal ill-defined myxoid areas containing vacuolated fibroblasts, suggestive of MIFS. However, other areas showed focal nuclear pleomorphism and a high mitotic index (18 mitoses per 10 high-power fields), possibly suggesting high-grade myxofibrosarcoma. 2 months later, the patient developed a recurrence at the site of the scar on the foot, which was confirmed on core biopsy. She was referred to our tertiary center where the tumor was resected. However, as there was tumor involvement of the excision margins in this specimen, the patient underwent a further excision 5 weeks later. While on follow-up, the patient was noted to have two local recurrences on the medial and lateral aspects of the dorsum of the right foot, 6 weeks following re-excision. At the same time, a raised pale lesion was noted on the dorsum of the right hand, which was also excised. The patient remains well and free of disease 6 weeks after her last treatment.
Results: pathologic findings
Grossly, the excision specimen of the recurrent tumor comprised an ovoid piece of skin measuring 80 mm × 62 mm and underlying subcutaneous tissue excised to a depth of 15 mm. The skin surface showed a 30 mm scar, and serial slicing revealed adipose tissue containing lobulated, heterogeneous tumor with gelatinous areas and multiple gray nodules. The re-excision specimen (taken 5 weeks later) was “C-shaped,” measuring 75 mm × 95 mm × 8 mm, and was covered by skin on one side. Areas of thickened skin were noted at the anterior-medial aspect and were sampled.
Histology from both specimens showed similar features of a dermal and predominantly subcutaneous, variably cellular, multinodular neoplasm (Figure 1(a)), comprising patternless distributions or sometimes more solid sheets of moderately atypical spindle cells and foci of vacuolated fibroblasts within prominent, although ill-defined, myxoid stroma with areas of hemorrhage (Figure 1(b)–(e)). In some areas, there were more cellular fascicles of atypical spindle cells infiltrating subcutaneous fat with some extension into the deep dermis. The mitotic index was up to 5 mitoses per 10 high-power fields, and no coagulative tumor necrosis was present. There was a patchy, relatively mild mixed inflammatory cell component. Lesional cells showed variable expression of CD34, but were negative for desmin, smooth muscle actin (SMA), S100 protein, and AE1/AE3. In addition, other areas showed nodules of adipose tissue with intervening, moderately cellular fibrous septa containing mildly atypical spindle cells with hemosiderin pigment in the cytoplasm (Figure 1(f) and 2(a)–(c)); the transition of these areas with the main tumor ranged from gradual to more abrupt (Figure 1(f)).
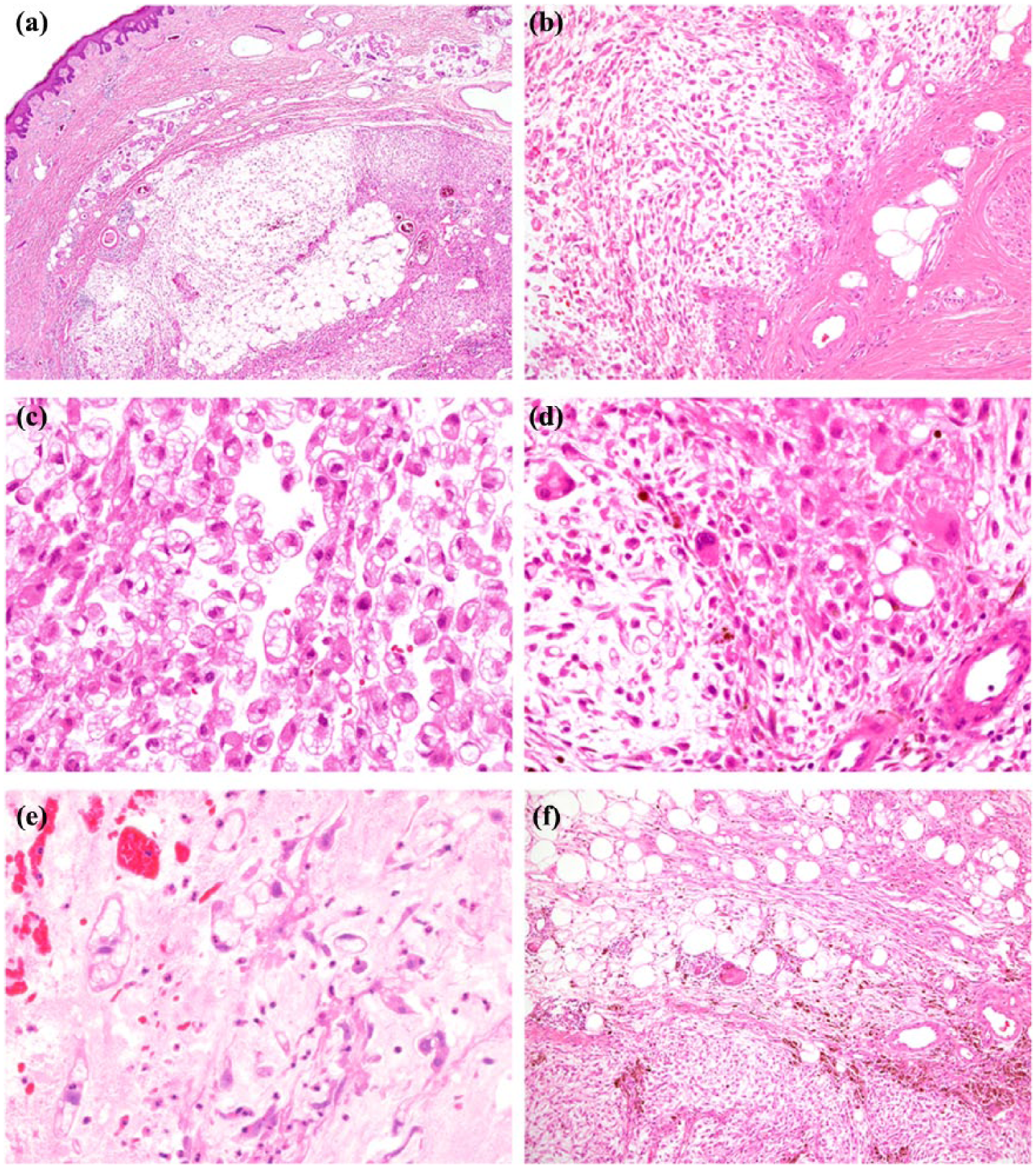
(a) Myxoinflammatory fibroblastic sarcoma (MIFS). This was a multinodular neoplasm centered in the dermal and subcutaneous tissues of the foot. (b–e) There are nodular distributions of numerous atypical, multivacuolated fibroblasts within extensive myxoid stroma (Figure 1(b)–(e)). Also present are small numbers of moderately atypical spindle cells and large cells with prominent enlarged eosinophilic nucleoli (Figure 1(d)). Foci of hemorrhage are present (Figure 1(e)). (f) Hemosiderotic fibrolipomatous tumor (HFLT). Many areas show adipose tissue with intervening, moderately cellular fibrous septa containing mildly atypical spindle cells with hemosiderin pigment deposition. The transition between the areas of HFLT and MIFS ranged from gradual to relatively abrupt (shown here).
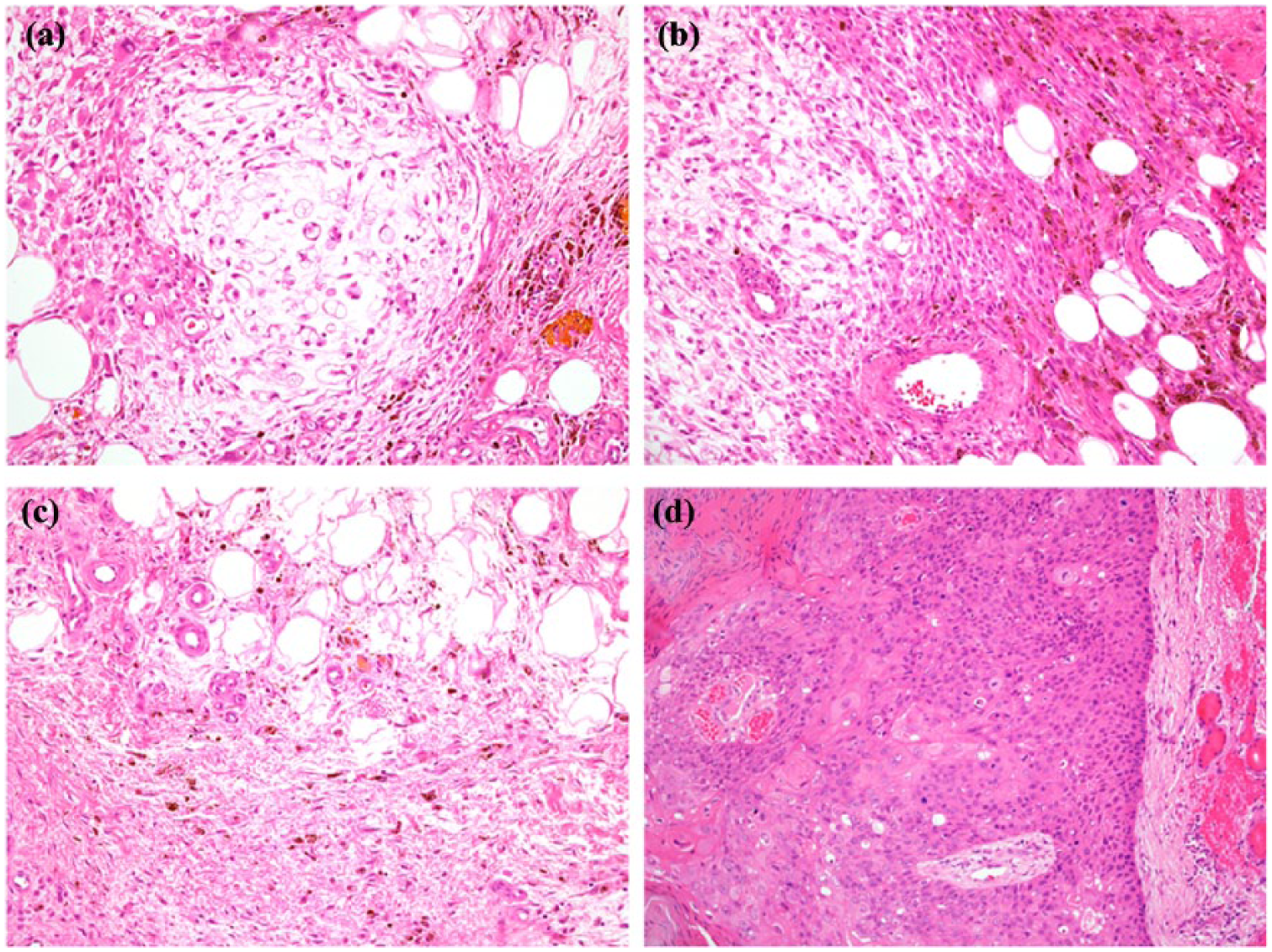
(a, b) HFLT and MIFS. The nodular and relatively well-demarcated areas of MIFS (left, Figure 2(a) and (b)) are seen adjacent to the fibrous septa of HFLT (right), which contain spindle cells and prominent hemosiderin pigment deposition. (c) HFLT. Sparsely cellular fibrous septa containing mildly atypical spindle cells intermingle with mature adipose tissue. (d) Bowen’s disease (squamous cell carcinoma in situ). The surrounding skin showed well-demarcated epidermal lesions comprising full thickness distributions of markedly atypical epithelial cells with atypical mitotic figures (Figure 2(d)), but without stromal invasion.
The features were consistent with MIFS and areas of HFLT. In the re-excision specimen, tumor was present at the deep margin and 4.5 mm from the nearest peripheral margin. The area of thickened skin at the anterior-medial aspect of the re-excision specimen showed squamous epithelium with full thickness atypia, including bizarre cells and atypical mitotic figures (Figure 2(d)). No stromal invasion was present. The features were of squamous cell carcinoma in situ (Bowen’s disease). The recurrent tumors on the right foot showed similar histologic features to the previous specimens, consistent with MIFS. However, features of HFLT were not seen in these specimens. The histology from the skin excision from the right hand showed multiple foci of Bowenoid actinic keratosis.
Discussion
We describe a case of MIFS of the foot of a 77-year-old female, which showed rapid and multiple local recurrences and arose in the setting of immune deficiency, 10 years after a renal transplant for polycystic kidney disease. MIFS is a rare malignant soft tissue neoplasm that typically occurs in middle-aged adults with a roughly equal gender distribution, and predominantly arises on extremity sites, particularly at the distal aspects. 2 MIFS was first described in 1998 as “inflammatory myxohyaline tumor” by Montgomery et al., 3 and by Meis-Kindblom et al. 1 who characterized it as “acral MIFS,” and by Michal 9 as “inflammatory myxoid tumor of the soft parts with bizarre giant cells.” While initially almost all tumors were reported in the distal extremities, “acral” was removed from the name after later reports of neoplasms at more proximal locations such as the thigh. 10 Most MIFSs behave as low-grade malignant neoplasms; although up to two-thirds have been noted to recur locally, 1 nodal and distant metastases are rare. The current treatment is wide local resection with clinical follow-up, although typically radiation therapy is indicated for multiply locally recurrent lesions and chemotherapy for patients with metastatic disease.
The etiology of MIFS is unknown, although some cases have been associated with histories of trauma to the lesion site. An infectious cause has been raised due to the presence of virocyte-like cells, but micro-organisms (including bacteria, fungi, herpes simplex virus, and cytomegalovirus) have been undetectable with histo- or immunohistochemistry. While Epstein-Barr virus (EBV) has occasionally been detected by polymerase chain reaction, these features were in keeping with latent infection only.2,3 The prominent inflammatory component seen in MIFS has led to the suggestion that mediators of chronic inflammation, including various cytokines, may be implicated in its development. 1
A history of transplantation has been reported only once, with a case of MIFS occurring as a nodular, subcutaneous dorsal foot mass in a 62-year-old male, who presented 10 months after renal transplantation for focal segmental glomerulosclerosis of unknown origin, and who had received triple immunosuppressive therapy with corticosteroids, cyclosporine, and mycophenolate mofetil. His donor’s serologies for infections and autoimmune diseases had all been negative (including EBV, cytomegalovirus, hepatitis B and C viruses, HIV, and syphilis), and he was reported as disease-free over 2 years after tumor excision. 11 Although this current case is only the second case of MIFS reported after transplantation, this highlights that immunocompromise (including the potential direct tumor-promoting effects of immunosuppressive therapies) 11 might be an etiologic factor.
Histologically, MIFS is characteristically a multinodular neoplasm comprising sparsely to moderately cellular distributions of three distinct cell types within prominent myxoid stroma containing a mixed inflammatory cell infiltrate, and the proportions of each cell type and of stromal and inflammatory components vary. The neoplastic cell types comprise first large pleomorphic “virocyte-like” cells with macronucleoli that resemble Reed–Sternberg cells; second, medium-sized to relatively large fibroblasts with cytoplasmic vacuolations containing extracellular mucinous material, sometimes leading to nuclear indentation and mimicking lipoblasts; and finally, moderately atypical spindle or epithelioid cells.1–3 While mitotic activity is usually low, some cases contain atypical mitoses or >10 mitoses per 10 high-power fields. The immunophenotype of MIFS varies, with about half expressing CD34 and CD68, but tumors typically being negative for SMA and epithelial membrane antigen. The behavior of MIFS cannot be reliably predicted from histologic parameters; cases with atypical features have been described by Laskin et al. 4 as containing (1) complex branching and/or thick-walled arcuate vessels in 20%–75% of the tumor; (2) small cellular areas of epithelioid or spindle cells in solid, fascicular, or whorled patterns; or (3) abnormal mitotic activity (>10 mitotic figures per 50 high-power fields and/or the presence of atypical mitoses). MIFS with at least two of these atypical features were noted to recur more frequently than those with only one feature or lacking these features; 4 despite this, complete initial surgical resection was the only statistically significant clinicopathologic factor for predicting a lower rate of recurrence. 4 This current case showed two of the criteria indicated, with both high mitotic index in the original specimen and more solid spindle cells areas, consistent with atypical MIFS according to this framework, and this may conceivably explain the rapid rate of local recurrence of this case.
HFLT is a benign lesion usually arising in the subcutis of the ankle or foot, particularly in women of middle age12,13 (with its predilection for distal extremity sites and most frequent occurrence in middle age shared with MIFS). HFLT is characterized by fibrohistiocytic-like spindle cells in septal and perivascular distributions (scattered cells often with enlarged hyperchromatic nuclei) admixed with mature adipose tissue, typically with honeycomb-like infiltration of adipocytic lobules, as well as hemosiderin deposition, hemorrhage and mixed inflammatory cells. This tumor has been associated with trauma or venous stasis, and can recur locally but has not been shown to metastasize. The lesional cells are positive for CD34 and calponin and sometimes for CD68 or lysozyme, but do not express other markers. Some lesions show hybrid features of MIFS and HFLT, with the two lesions either being discretely demarcated, or with more intermingled or transitioning features.14,15
Genetically, MIFS has been associated with a marker/ring chromosome 3 with 3p amplicons 16 as well as with a characteristic translocation, t(1;10)(p22: q24) with rearrangements of TGFBR3 and MGEA5 on chromosomes 1p22 and 10q24. These gene rearrangements are shared with HFLT 17 and pleomorphic hyalinizing angiectatic tumor, 18 and it was postulated that these neoplasms could represent a clinicopathologic spectrum. However, these abnormalities have not been found in all cases of MIFS.18,19 Antonescu et al. 15 documented consistent t(1;10) with TGFBR3 and MGEA5 gene rearrangements in both MIFS and HFLT, including all three hybrid MIFS/HFLT studied, suggesting that these might represent different clinical and morphologic spectra of the same biologic entity. TGFBR3 and/or MGEA5 rearrangements have also been shown in 3/3 hybrid MIFS/HFLT by Carter et al. 18 However, this group recently assessed a large series including MIFS, HFLT and MIFS and found only 2/31 MIFS to contain MGEA5 rearrangements with no MIFS harboring TGFBR3 rearrangements, while 1/1 HFLT showed TGFBR3 and MGEA5 rearrangements and 6/8 hybrid HFLT/MIFS contained TGFBR3 and/or MGEA5 rearrangements. 20 These authors therefore postulated that because TGFBR3 and MGEA5 rearrangements were more frequent in hybrid HFLT/MIFS than in classic MIFS, MIFS and HFLT may not be related, and that hybrid HFLT/MIFS could represent HFLTs showing progression to sarcomatous features rather than HFLT and MIFS indicating a single entity. 20
The differential diagnosis of MIFS varies according to the proportion of tumor cell, inflammatory cell and stromal components of each case, and diagnosis may be particularly challenging in limited biopsy material, so awareness of the likelihood of morphologic variation is required. If more sparsely cellular myxoid zones are sampled, the neoplasm can resemble myxoma or cellular myxoma. If a prominent inflammatory component is sampled, this can resemble an inflammatory lesion or metastatic tumor in lymph node, and prominent virocyte-like/Reed-Sternberg cells can mimic Hodgkin lymphoma. Myxofibrosarcoma also typically arises on the extremities of older patients and is a multinodular myxoid neoplasm, but instead contains one cell type, of variably atypical spindle to epithelioid cells, rather than the three characteristic cell types of MIFS. In addition, myxofibrosarcoma is associated with prominent large curvilinear vessels dispersed within the myxoid stroma and, although inflammatory cells may be present, this is not the marked mixed inflammatory cell infiltrate seen in MIFS. Myxoid liposarcoma (MLPS) also contains prominent myxoid stroma and typically arises at extremity sites, but it predominantly occurs in younger adults and at more proximal extremities. MLPS comprises patternless distributions of bland ovoid and spindle cells without cellular atypia dispersed in lightly basophilic myxoid stroma that contains small, delicate curvilinear vessels that are lacking in MIFS. MLPS may also contain lipoblasts with indented nuclei, in contrast to the “pseudolipoblasts” of MIFS that contains mucinous material rather than fat. Genetically, MLPS harbors characteristic FUS–DDIT3 or, more rarely, EWSR1–DDIT3 gene fusions.
In summary, this is a case of MIFS occurring in an immune-deficient patient. This case gives further insight into the clinical setting of this rare soft tissue neoplasm and highlights immunocompromise/impaired immune surveillance as a possible factor in its development. While this neoplasm shares similar features to other sarcomas such as myxofibrosarcoma and undifferentiated pleomorphic sarcoma, it is important to recognize its specific features to enable correct prognostication and, because of the distinct genetic background of MIFS, to realize the potential for specific targeted therapies in future.
Footnotes
Acknowledgements
The authors acknowledge support from the NIHR Royal Marsden/ICR Biomedical Research Center. M.H., Y.M., A.J.H., R.L.J., C.F., and K.T. obtained clinical information; M.H., Y.M., C.F., and K.T. wrote the manuscript; K.T. prepared the images.
Conflict of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Full informed consent to publish manuscript and images obtained from the patient.