
Abstract
Giant lymph node hyperplasia is the main symptom of Castleman’s disease (CD), which is a rare and easily overlooked lymphoproliferative disease that mimics both benign and malignant lesions. Several cases of CD after renal transplantation have been reported in the literature. A 43-year-old man was admitted to our medical center with high serum globulin levels after receiving a living donor kidney transplant. A lymph node biopsy raised suspicion for multicentric CD. Because of the poor therapeutic effect of reduction of immunosuppression, the patient was treated with CHOP chemotherapy comprising four cycles of monthly cyclophosphamide (750 g/m2, day 1), vincristine (1.4 g/m2, day 1), doxorubicin (50 g/m2, day 1), and prednisone (60 mg/m2, daily). Following the treatment, his serum globulin levels decreased to the normal range. At the 2-year follow-up examination, the abdominal, axillary, and inguinal lymph nodes had significantly decreased in size. Without a pathological diagnosis, multicentric CD after renal transplantation can be easily ignored and misdiagnosed. If the clinical signs cannot be relieved by minimizing the dose of common immunosuppressants, the CHOP regimen may be a better option. Biological agents may be added in patients with positive immunohistochemistry staining and good economic conditions.
Keywords
Background
Solid organ transplantation (SOT) and hematopoietic stem cell transplantation (HSCT) have rescued thousands of patients. In recent years, however, post-transplant lymphoproliferative disorders (PTLDs) have been observed in some patients who have undergone SOT or HSCT. A PTLD is defined as a disease of lymphocyte dysplasia caused by immune dysfunction after HSCT or SOT. Giant lymph node hyperplasia is the main symptom of Castleman’s disease (CD), which is a rare and easily overlooked lymphoproliferative disease that mimics both benign and malignant lesions. CD is primarily associated with viruses, such as Epstein–Barr virus (EBV), human immunodeficiency virus (HIV), or human herpesvirus 8 (HHV-8). Because of the onset of occult disease and lack of specificity, the diagnosis of CD requires pathological biopsy, and it is easily missed and misdiagnosed. Although the incidence of CD after kidney transplantation is low, CD often leads to the development of malignant PTLDs such as B-cell lymphoma and T-cell lymphoma. The development of CD after renal transplantation has been previously described only twice. To our knowledge, this is the first reported case of a renal transplant recipient maintained with tacrolimus and mycophenolate mofetil. The main manifestations were a persistently high serum immunoglobulin level and generalized lymphadenopathy. EBV and cytomegalovirus (CMV) infection after transplantation is associated with neoplastic and non-neoplastic diseases. CD is a rare complication but should be taken into consideration in patients who have undergone SOT. We herein present the above-mentioned case and further review the clinical features, pathological features, and treatment of CD after kidney transplantation to improve clinicians’ awareness (Tables 1 and 2).
Review of case reports of Castleman’s disease in SOT recipients: demographic data and onset characteristics.

SOT, solid organ transplantation; CMV, cytomegalovirus; HHV-8, human herpesvirus 8; EBV, Epstein–Barr virus; IL-6, interleukin 6.
Review of case reports of Castleman’s disease in SOT recipients: clinical findings.
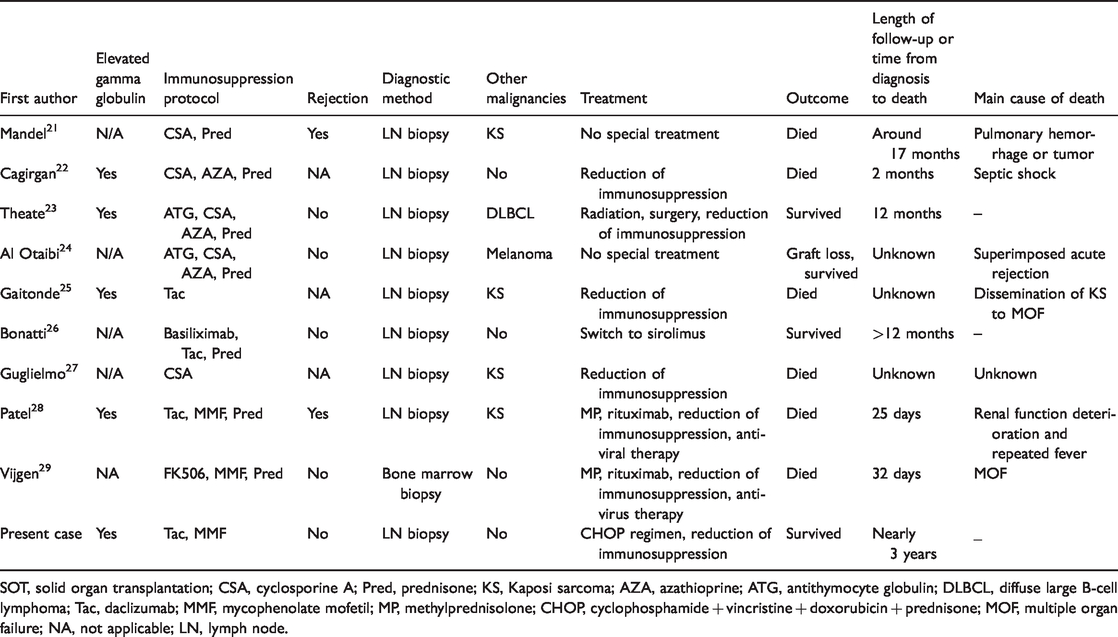
SOT, solid organ transplantation; CSA, cyclosporine A; Pred, prednisone; KS, Kaposi sarcoma; AZA, azathioprine; ATG, antithymocyte globulin; DLBCL, diffuse large B-cell lymphoma; Tac, daclizumab; MMF, mycophenolate mofetil; MP, methylprednisolone; CHOP, cyclophosphamide + vincristine + doxorubicin + prednisone; MOF, multiple organ failure; NA, not applicable; LN, lymph node.
Case presentation
A 43-year-old man received an ABO-compatible living donor kidney transplant donated from his father in July 2012 for treatment of end-stage renal disease. The panel-reactive antibody test of the recipient and crossmatch were negative with two mismatches of HLA-A and HLA-DQ. Neither the recipient nor donor had a history of CD. Serum antibodies to HIV, EBV, and CMV were negative before the operation. The recipient received induction therapy with a 14-day course of antilymphocyte globulin (Pressimum; Behringwerke AG, Marburg, Germany). The immunosuppressive therapy started 3 days prior to transplantation. The immunosuppressive regimen consisted of tacrolimus at 3.5 mg twice daily, mycophenolate mofetil beginning at 750 mg twice daily and tapered off during a 4-week period, prednisone beginning at 20 mg daily and tapered off during a 6-week period, and valganciclovir at 450 mg daily for 3 months post-transplantation. The prednisone was withdrawn in the second month after transplantation because of gastrointestinal bleeding. At 4 years post-transplantation, the serum globulin level was found to have gradually increased without any patient discomfort. Physical examination revealed no abnormalities. The serum creatinine level was maintained at a normal level, fluctuating from 1.1 to 1.2 mg/dL, and the urine protein was negative. The white blood cell count was mildly elevated at 10,580 cells/mm3 (normal range, 4000–10,000 cells/mm3) with 61.5% neutrophils. Serum EBV and CMV immunoglobulin (Ig)M and IgG antibodies were positive, while serum HIV and HHV-8 antibodies were negative. The RNA qualification of EBV showed 4.6 × 104 copies/mL, and CMV-DNA was negative. The serum level of interleukin 6 (IL-6) was normal. B-mode ultrasonography and computed tomography showed multiple lymphadenectases, including the bilateral cervical, inguinal, and abdominal cavity lymph nodes. No visceral enlargement or bone marrow involvement was found. Furthermore, positron emission tomography showed no abnormal metabolic signals of organomegaly or extranodal masses. Serum protein electrophoresis showed a high serum free light chain κ/λ level and low κ/λ ratio. Serum protein electrophoresis also showed an elevated percentage of gamma globulin but did not identify the monoclonal gammopathy. Immunofixation electrophoresis showed M protein negativity (Table 3).
Results of protein electrophoresis during follow-up.

The patient underwent biopsy of the right inguinal lymph node in a local hospital. The pathologic examination revealed a follicular pattern with some hyperplastic germinal centers, most of which were small and similarly sized with a round or oval shape. Numerous mature plasma cells were seen in medullary cords (Figure 1(a)). Cohesive large cells were present mainly in sinuses, and monocytoid B-cell proliferation was scattered in the mantle zone of the follicles; these B cells exhibited features of immunoblasts or anaplastic large cells (Figure 1(b)). Focally, the architecture was entirely destroyed by sheets of large typical cells. The pathological diagnosis was reactive lymphoid tissue of proliferated lymph nodes and slightly increased plasma cells increased in the medulla of the lymph nodes. Immunohistochemistry showed positive staining of CD3, CD5, CD43, CD38, CD138, CD21, CD23, CD20, CD79, BCL2, and Ki-67 (<10%) in the large cells, with negative staining of CK, CD10, CD56, cyclin D1, SOX11, and IgG4. EBV-encoded early RNAs were found in the plasma cells based on in situ hybridization, while CMV inclusion bodies were not observed.

Immunohistochemical staining of lymph nodes from biopsy. (a) Reactive lymphoid tissue hyperplasia in the left inguinal lymph nodes. (b) Plasma cell proliferation in the abdominal lymph nodes. (c–h) Positive stains in the abdominal lymph nodes, including CD3, CD20, CD38, CD138, kappa light chains, and lambda antibodies.
Because of the persistently elevated immunoglobulin level and generalized lymph node enlargement, another celiac lymph node biopsy was performed. Pathological evaluation of the celiac lymph node showed that multiple lymphoid follicles had been infiltrated by a large number of mature plasma cells. A few small blood vessels showed hyaline degeneration with “onion skin” like changes. The germinal centers were atrophied, and the proliferation of the sleeve cells and concentric round arrangement were not obvious. The pathological diagnosis was the characteristic plasma cell subtype of CD. The plasma cells were positive for CD3, CD20, CD38, CD138, and Ki-67 (<10%) (Figure 1(c)–(f)). CD138 showed strong positive expression in the plasma cells labeled by polyclonal anti-immunoglobulin kappa light chains and lambda antibodies (Figure 1(g), (h)). Immunostaining for EBV and CMV was negative. We thereafter adjusted the dosage of immunosuppressants, adding prednisolone at 10 mg once a day, slowly tapering the tacrolimus down from 3.5 mg twice daily to 0.5 mg twice daily, and maintaining the mycophenolate mofetil at 500 mg twice daily. Two months after adjustment of the immunosuppression agents, the elevated immunoglobulin level and enlarged lymph nodes were not ameliorated. Because of the patient’s economic conditions and risk of infection, he did not receive rituximab to treat the multicentric CD. Instead, he received a CHOP chemotherapy regimen consisting of monthly cyclophosphamide (750 g/m2, day 1), vincristine (1.4 g/m2, day 1), doxorubicin (50 g/m2, day 1), and prednisone (60 mg/m2, once daily) for four cycles. After four cycles of chemotherapy, the enlarged lymph nodes had significantly decreased in size (Figure 2). At the third month of follow-up, the serum globulins had dropped to a normal level (Figure 3). At the ninth month of follow-up, the size of the lymph nodes located in the abdomen, axilla, and groin had significantly decreased. No acute rejection occurred during the post-transplantation follow-up. The serum creatinine level was maintained within the normal range, fluctuating from 1.1 to 1.2 mg/dL.
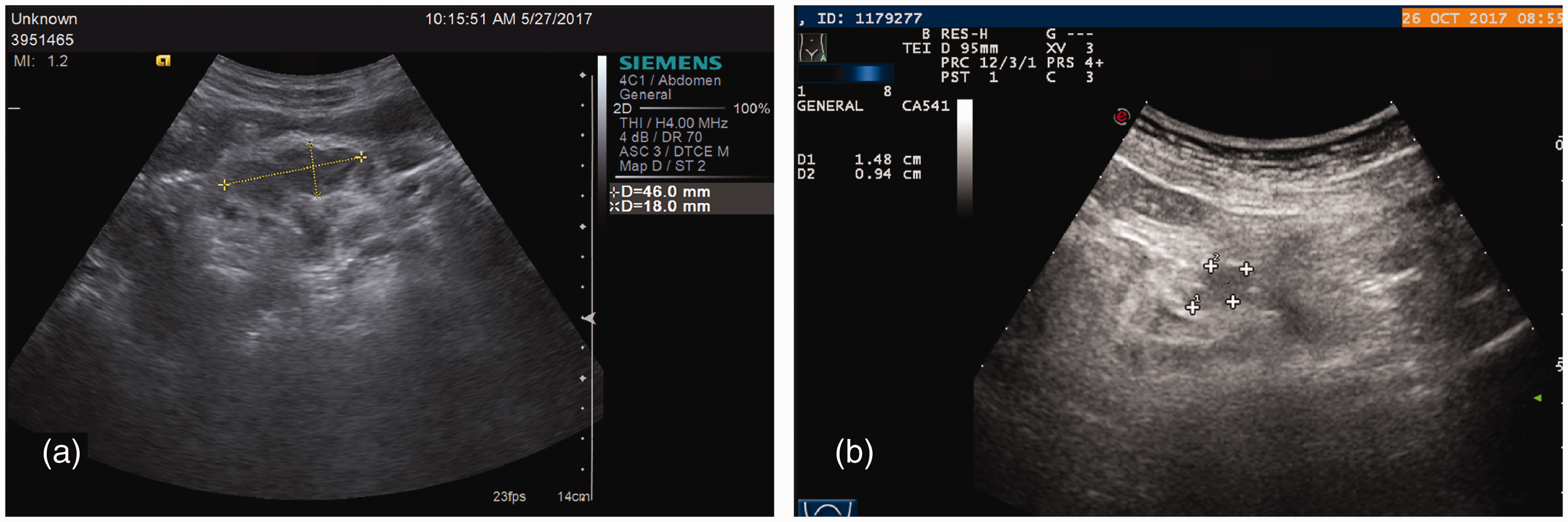
Size of the abdominal lymph nodes detected by ultrasound examination. (a) An enlarged left inguinal lymph node with a diameter of 46 × 18 mm. (b) The left enlarged inguinal lymph node shrank after treatment, decreasing to a diameter of 15 × 9 mm.

Dynamic changes of serum immunoglobulins before and after treatment. Ig, immunoglobulin
Discussion
With more than 28,000 SOTs performed annually in the United States, SOT for individuals with end-stage organ disease is a very important life-saving option. 1 To our knowledge, patients undergoing SOT routinely receive intensive long-term immunosuppressive therapy to prevent graft rejection, which puts them at high risk of opportunistic infections and de novo malignancies. PTLDs are a rare but potentially fatal complication. In 1968, Doak et al. 2 described the first case of a PTLD in a renal transplant recipient. Since then, a large amount of information has been gathered on risk factors, diagnosis, classification, treatment, and prognosis. However, PTLDs are is still highly heterogeneous, characterized by a series of pathological and clinical manifestations ranging from benign lymphoid hyperplasia to invasive lymphoma.
In 1956, CD was first described as benign giant lymphoma, angiofollicular lymph node hyperplasia, and follicular lymphoblastoma. 3 CD has been divided into two subtypes according to its prognosis: unicentric CD and multicentric CD. In addition, CD is believed to be commonly associated with non-Hodgkin’s lymphoma, Hodgkin’s lymphoma, and Kaposi sarcoma. 4 The main clinical manifestations of multicentric CD are fever, anemia, fatigue, lymphadenopathy, and hypoproteinemia. The typical presentations are generalized lymph node enlargement and splenomegaly, which were observed in the present case and in previous reports. CD may be easily misdiagnosed as lymphoma. 5 The histopathological characteristics of the lymph nodes in patients with CD include immune cell aggregation, germinal center hyperplasia, plasma cell proliferation, and vascular proliferation. The serum IL-6 level is associated with symptom severity, lymph node lesion characteristics, and lymph node size. 6 Elevated inflammatory markers such as IL-6 and C-reactive protein are commonly detected in the serum.7,8 The lymph nodes of patients with CD show an increased level of IL-6 without an increase in the expression of other cytokines. Matsunami et al. 9 reported that tocilizumab, a humanized monoclonal blocker for the IL-6 receptor, is effective for multicentric CD and ensures a safe perioperative period during kidney transplantation. However, among all cases in our literature review, only one had a high IL-6 level.
The eight previously reported cases of adults who developed multicentric CD post-transplantation involved five renal recipients, two post-liver transplant patients, and one heart transplant recipient. The interval from transplantation to the onset of CD ranged from 6 months to 16 years. Four patients had concomitant multicentric CD and Kaposi sarcoma, all of whom died, and these two conditions were more easily diagnosed in HHV-8 endemic regions. The diagnosis of multicentric CD and KS was confirmed by lymph node biopsy at the same site in three patients, while one patient was diagnosed at different sites.
PTLDs are also believed to be associated with EBV infection. 10 Because the function of effector T lymphocytes is inhibited by various factors after transplantation, EBV-infected B lymphocytes grow out of control, resulting in abnormal lymphocyte hyperproliferative diseases or monoclonal malignant tumors. Multicentric CD is often associated with HIV or HHV-8 in patients undergoing SOT. 5 However, other infections were present among five of the eight adult patients in our study, including cholangitis, chronic urinary tract infection, and chronic hepatitis C. Notably, only one patient had acute cellular rejection, suggesting that exposure to HHV-8/EBV instead of strengthening of immunosuppression may be a key risk factor. A recent study suggested that there is a high risk of serum HHV-8 conversion among Italian transplant recipients. 11 Of the eight reported adult patients, six had a history of other malignant tumors, including Kaposi sarcoma (n = 4), EBV-related PTLD (n = 1), and melanoma (n = 1). HHV-8 negativity was detected in both our patient and his organ donor. However, neither the immunohistochemical results nor the HHV-8 viral load data were reported in any of the previously published cases.
Different treatment options are adopted according to the subtype of CD. For unicentric CD, most clinicians recommend resection of enlarged lymph nodes. A better prognosis and low recurrence rate can be achieved. 12 Treatment of multicentric CD includes immunosuppressant reduction, chemotherapy, and immunotherapy. Antiretroviral therapy is performed when HIV/HHV-8 positivity is confirmed. Monotherapy with an IL-6 inhibitor is preferred over rituximab and other immunotherapy drugs in patients with HIV or HHV-8 negativity, though the etiology and pathophysiology of HHV-8–negative multicentric CD are unknown. 13 Cytotoxic chemotherapy regimens, mainly CHOP, are effective in most severe cases by removing a large number of cytokine-secreting cells, but recurrence is common and adverse effects are significant.14,15 Despite the high cost of immunotherapy, recent advances have shown that rituximab improves the treatment efficacy in patients with Kaposi sarcoma-associated herpesvirus multicentric CD and reduces the need for cytotoxic chemotherapy. 16
A case involving a 56-year-old man with CD who improved after kidney transplantation was recently reported. 17 Corticosteroids inhibit inflammation and reduce lymphocyte proliferation. Thus, these drugs are an option for relieving symptoms during acute exacerbations of idiopathic multicentric CD; however, most patients relapse when the steroid dose is gradually decreased. 18 Sirolimus, a proliferation signal inhibitor, is suggested to induce the remission of multicentric CD by converting immunosuppressive schemes from the calcineurin inhibitor. 19 However, Ariza-Heredia and Razonable 20 reported that a high concentration of sirolimus is associated with the development of Kaposi sarcoma with a mortality rate ranging from 8% to 14%.
In the absence of any standard effective treatment for this syndrome, the CHOP chemotherapy regimen was applied in the present case, and sustained regression was induced following four cycles of chemotherapy. The immunoglobulin level decreased to the normal range 2 months after chemotherapy and was still stable at the 2-year follow-up. In our literature review, six patients who had been treated with rituximab or other biologics died. Whether the use of rituximab was associated with the clinical deterioration of these previously reported patients remains unclear. 8 The use of anti-IL-6 drugs may result in better outcomes, as proposed by some researchers. 16 Because a few patients described in the literature were diagnosed with a combination of Kaposi sarcoma and multicentric CD, there is no definite conclusion on the most effective treatment for this syndrome. 13 Further research on the pathogenesis of multicentric CD is needed to identify additional candidate targeted therapies.
Conclusion
Multicentric CD after renal transplantation must rely on a pathological diagnosis because it can be easily missed or misdiagnosed. Tapering of traditional immunosuppressive agents has a poor prognosis, while the CHOP regimen can obtain a better curative effect. Biological agents may be added in patients with positive immunohistochemistry staining and good economic conditions.
Footnotes
Acknowledgements
The authors would like to thank Gio Leedy and Xue Xing for their invaluable assistance.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This research was supported by funding from the National Natural Science Foundation of China (8170752).