
Abstract
Accurate detection of epileptogenic lesions on imaging requires a systematic search for abnormalities with diverse etiologies, appearances, and locations. This article provides a practical approach to the detection of epileptogenic foci for radiologists. First, we review protocols tailored to seizure evaluation. Next, we present a pictorial review of structural etiologies for seizure, including temporal lobe pathologies, developmental malformations, neoplastic lesions, vascular lesions, and infectious/inflammatory processes. We provide a step-by-step, systematic approach to ensure detection of the manifold potential pathologies visible on imaging as well as appropriate correlation with clinical findings. Finally, we present example reporting templates which can ensure efficient reporting by radiologists as well as serve as mnemonic aids.
Keywords
Introduction
Epilepsy is a neurological disorder in which a person has a risk of recurrent seizures due to a chronic underlying process. 1 Epilepsy is diagnosed after 2 recurrent unprovoked seizures occurring more than 24 hours apart, after a single unprovoked or reflex seizure with a high risk of reoccurrence (at least 60% risk over the next 10 years), or diagnosis of an epilepsy syndrome.2,3 The incidence of epilepsy in the U.S. is 79 cases per 100,000 persons with a prevalence of 8 persons per 1000. 4 Epilepsy exists in many forms with multiple causes encompassing structural, genetic, infectious, metabolic, immune, and unknown etiologies.5,6 An important aspect of diagnostic evaluation for epileptic seizure is classification based on seizure type: focal, generalized, or unknown.2,6
Structural causes of epilepsy grouped by imaging abnormality.
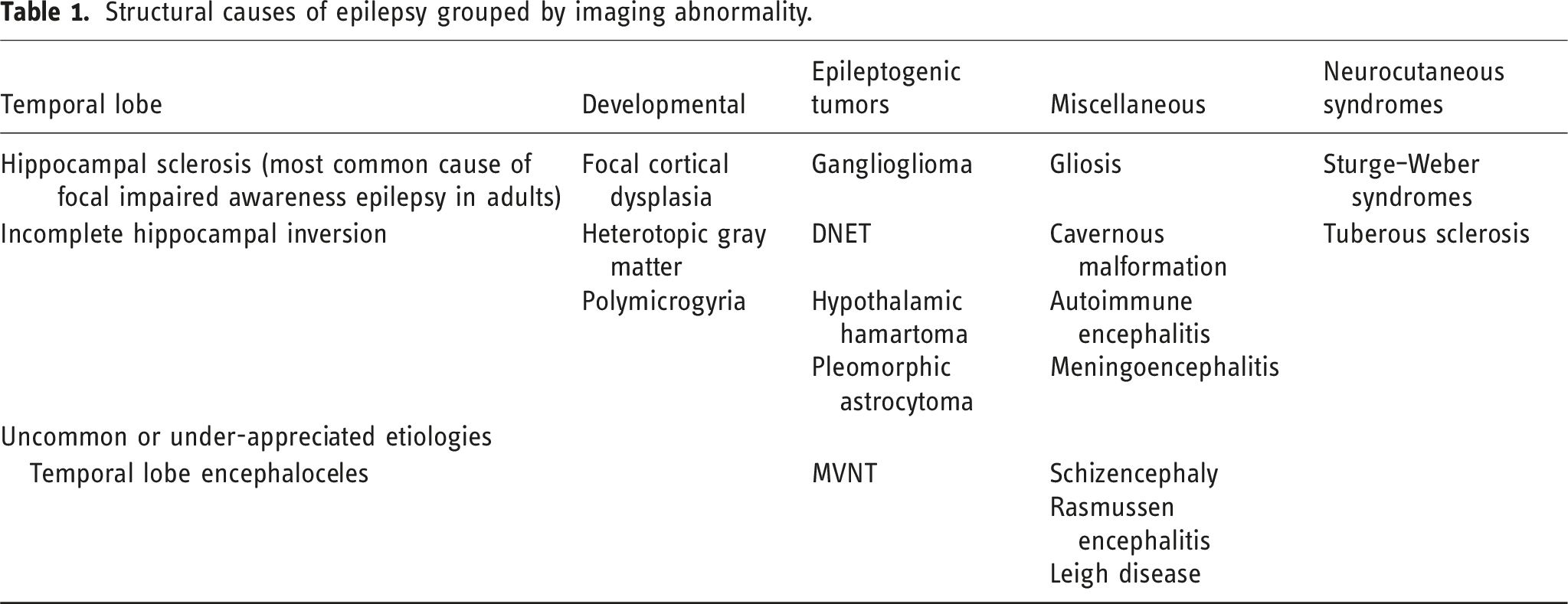
In the nonemergent setting, neuroimaging with MRI using specialized seizure or epilepsy protocols is performed for evaluation of structural brain lesions such as infarction, infection, trauma, tumors, vascular malformation, developmental abnormalities, and other seizure-associated brain pathologies. 9 In the emergent setting, CT is the initial modality used to rapidly identify immediately treatable structural pathologies while retaining ready access to the patient.5,9,10 The presence of any of these entities must then be further evaluated with integration of clinical semiology and electroclinical studies by the epileptologist to establish a causal relationship between the structural lesion and the patient’s epilepsy. Identification of an epileptogenic lesion on MRI should prompt referral to a specialized epilepsy surgery center for surgical evaluation as the presence of a structural lesion is a strong indicator of anti-epileptic drug resistance. 10 Furthermore, small lesions identified by MRI and electroclinical studies may be precisely targeted within highly eloquent regions using minimally invasive procedures such as stereotactic radiofrequency thermoablation and newer stereotactic laser ablation.11,12
Imaging protocol/advanced techniques
Specialized MRI epilepsy protocols are crucial for detection of various structural pathologies that may be missed by routine MRI.9,13 The ILAE Neuroimaging Task Force identified a set of sequences known as HARNESS-MRI, consisting of high-resolution 3D T1-weighted gradient echo sequence (no interslice gap) with isotopic voxels of 1 mm or less for optimal evaluation of brain anatomy and morphology, high-resolution 3D fluid-attenuated inversion recovery (FLAIR) turbo spin echo sequence (no interslice gap) with isotropic millimetric voxel resolution for assessing signal anomalies, and high in-plane resolution 2D coronal T2WI turbo spin echo sequence with submillimetric voxel resolution (e.g., 0.4 × 0.4 × 2 mm3, no interslice gap) for assessing hippocampal internal structure, amygdala, and parahippocampal cortices. 10 An important caveat for FLAIR is that it is not sensitive for epileptogenic lesions in children before 24 months due to incomplete myelination. 10 Additional MRI sequences include T1WI with gadolinium for evaluation of suspected tumor, vascular malformation, or infectious process and susceptibility-weighted imaging for detection of venous blood, hemorrhage, iron deposits, and calcifications. 10 Several of the entities discussed in this review, such as hippocampal sclerosis and cortical malformations, are better appreciated on scanners with magnetic fields of 3T or higher due to increased signal-to-noise ratio and resolution. 13
Despite use of these specialized epilepsy protocols, patients with epileptogenic foci may still have visually normal MRI, which has prompted efforts to develop advanced neuroimaging sequences. 14 Double inversion recovery (DIR) is a newer MRI structural protocol with high contrast between gray and white matter for better detection of epileptogenic lesions compared to FLAIR, T1WI, and T2W2 in temporal lobe epilepsy (TLE) and extratemporal focal epilepsy. 14 Fluid and white matter suppression (FLAWS) is a technique that suppresses white and cerebrospinal fluid signals to generate gray matter-specific images for better detection of focal cortical dysplasia. Edge-enhancing gradient echo (EDGE) is another technique for focal cortical dysplasia that directly visualizes the boundary between gray and white matter. 14
Epileptogenic entities
A wide spectrum of etiologies may produce seizures, only a subset of which can be detected via imaging. Non-structural factors contributing to seizures include metabolic derangements, channelopathies, and genetic syndromes. Structural abnormalities identifiable on neuroimaging include temporal lobe pathologies, developmental malformations, neoplastic lesions, vascular lesions, as well as infectious and inflammatory processes. Even when structural abnormalities are present, it is crucial to incorporate electrophysiological information and to pinpoint the abnormal structures that are truly epileptogenic foci.
Temporal lobe etiologies
Hippocampal sclerosis (HS), also known as mesial temporal sclerosis, is the most common cause of medically refractory epilepsy.
13
The underlying pathophysiology characterized by hippocampal neuronal loss and gliosis. In patients undergoing epilepsy surgery, HS was identified within specimens in 44% of adults and 15% of children.
15
Mesial temporal lobe epilepsy syndrome is associated with focal seizures with impaired consciousness, history of febrile seizures, family history of epilepsy, early onset, and intractable seizures not responsive to anti-epileptic medications. Findings on imaging may be bilateral but are usually unilateral or asymmetric.
9
Characteristic features include hippocampal volume loss or atrophy, abnormal T2 hyperintensity, and obscuration of internal architecture (Figure 1). Secondary signs include ipsilateral fornix and mammillary body atrophy, as well as enlarged temporal horn and choroidal fissure.
16
Use of 3T, or higher, scanners enables quantitative assessment of hippocampal volume and visualization of hippocampal internal architecture.
13
Furthermore, use of DIR has been reported to have superior detection of temporal white matter abnormalities compared to FLAIR.
14
Abnormal findings should be integrated with clinical semiology and electrophysiological findings to identify the epileptogenic focus. Hippocampal sclerosis: Volumetric brain analysis (a) and coronal FLAIR (b) of a 36-year-old male with new-onset seizures showing asymmetrically reduced volume of the left hippocampus (oval) in comparison to the right with associated increased FLAIR signal compatible with hippocampal sclerosis (circle).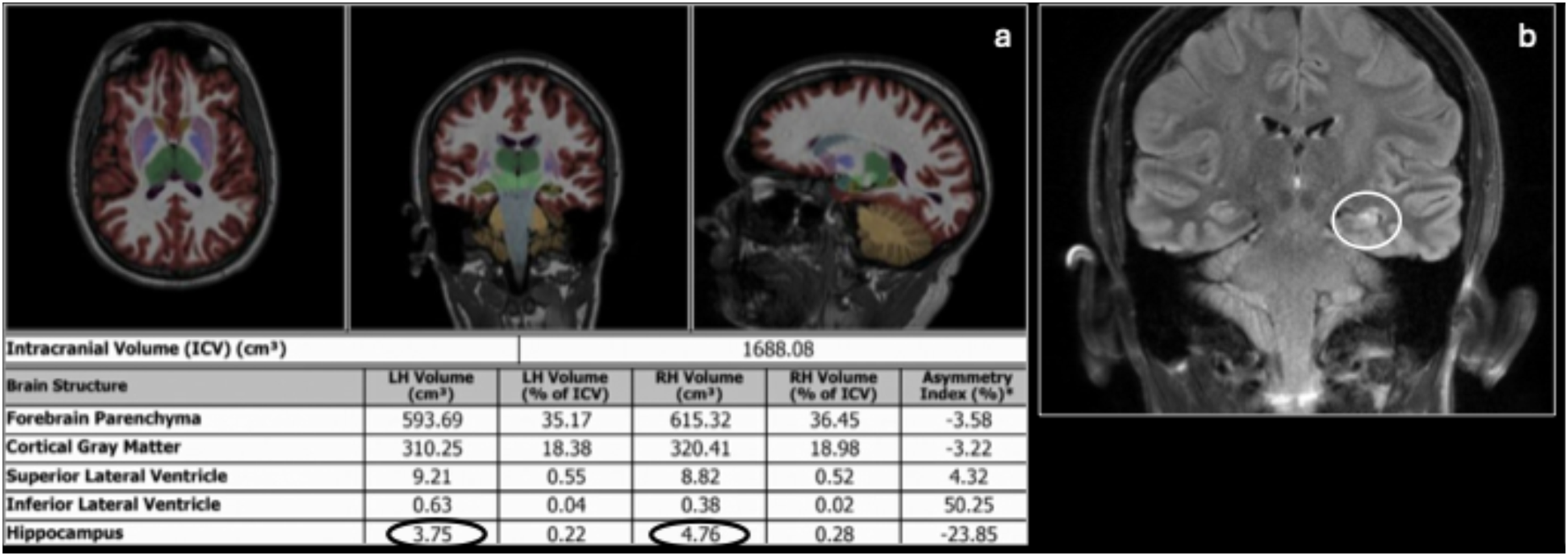
Developmental etiologies
Focal cortical dysplasia
Malformations of cortical development (MCD) are a group of conditions, often with childhood onset, caused by abnormal cell proliferation or apoptosis, abnormal neuronal migration, or abnormal cortical organization. 13 Focal cortical dysplasia (FCD) is the most common MCD responsible for extratemporal epilepsy. Prevalence of FCD ranges from 5 to 25% depending on collection and imaging technique. 17 FCD is present in 5–10% of epilepsy patients and has a prevalence of up to 50% in children with intractable focal epilepsy. 13 Despite its prevalence in intractable epilepsy, it is potentially curable by surgery.
FCD is classified into types and subtypes with varying degrees of abnormalites.
18
FCD type I is characterized by mild cortical dyslamination without dysmorphic neurons and presents with absent or subtle MRI findings, such as mild focal cortical thickening, asymmetrical gyral pattern, and blurring of the gray–white matter junction. Type IIa is characterized by cortical lamination with dysmorphic neurons and is more reliably identified with MRI, presenting with cortical thickening, blurred gray–white matter junction, and hyperintense cortical and subcortical signals. Type IIb presents with even more pronounced imaging findings such as cortical thickening, marked blurring of the white–gray matter junction, and a highly specific but not always present transmantle sign or radial band that appears as T2 hyperintense wedge-shaped signal in the subcortical white matter (Figure 2).18,19 FCD type III is characterized by features of FCD type I or type II in association with other lesions, such as HS (type IIIa), tumors (type IIIb), vascular malformations (type IIIc), or acquired lesions (IIId). Focal cortical dysplasia: Axial (a) and coronal (b) FLAIR and T1 (c) MR images of a 20-year-old female with new-onset of seizures show a region of abnormal high FLAIR signal (circle) with associated T1 hypointensity (circle) in the left middle temporal gyrus with associated blurring of the gray/white matter interface, cortical thickening, and a subtle transmantle sign (arrow) suggestive of FCD type IIb. Axial FLAIR (d) MRI from a 32-year-old female shows an incidental region of faint cortical thickening (circle) and FLAIR hyperintensity (arrow) measuring approximately 15 mm in the posterior superior margin of the left middle frontal gyrus, suggestive of FCD type IIa. Stability of the lesion was demonstrated on multiple follow-up exams. Axial (e) and sagittal FLAIR (f) MRI showing asymmetric thickening (oval) of the right anterior superior and middle frontal gyrus with blurring of the gray–white matter junction (circle) and subtle hyperintensity on FLAIR. Color-coded FDG-PET (g) in the same patient shows focally decreased metabolism in the right frontal lobe (arrows), correlating with the region of focal cortical dysplasia noted on MRI.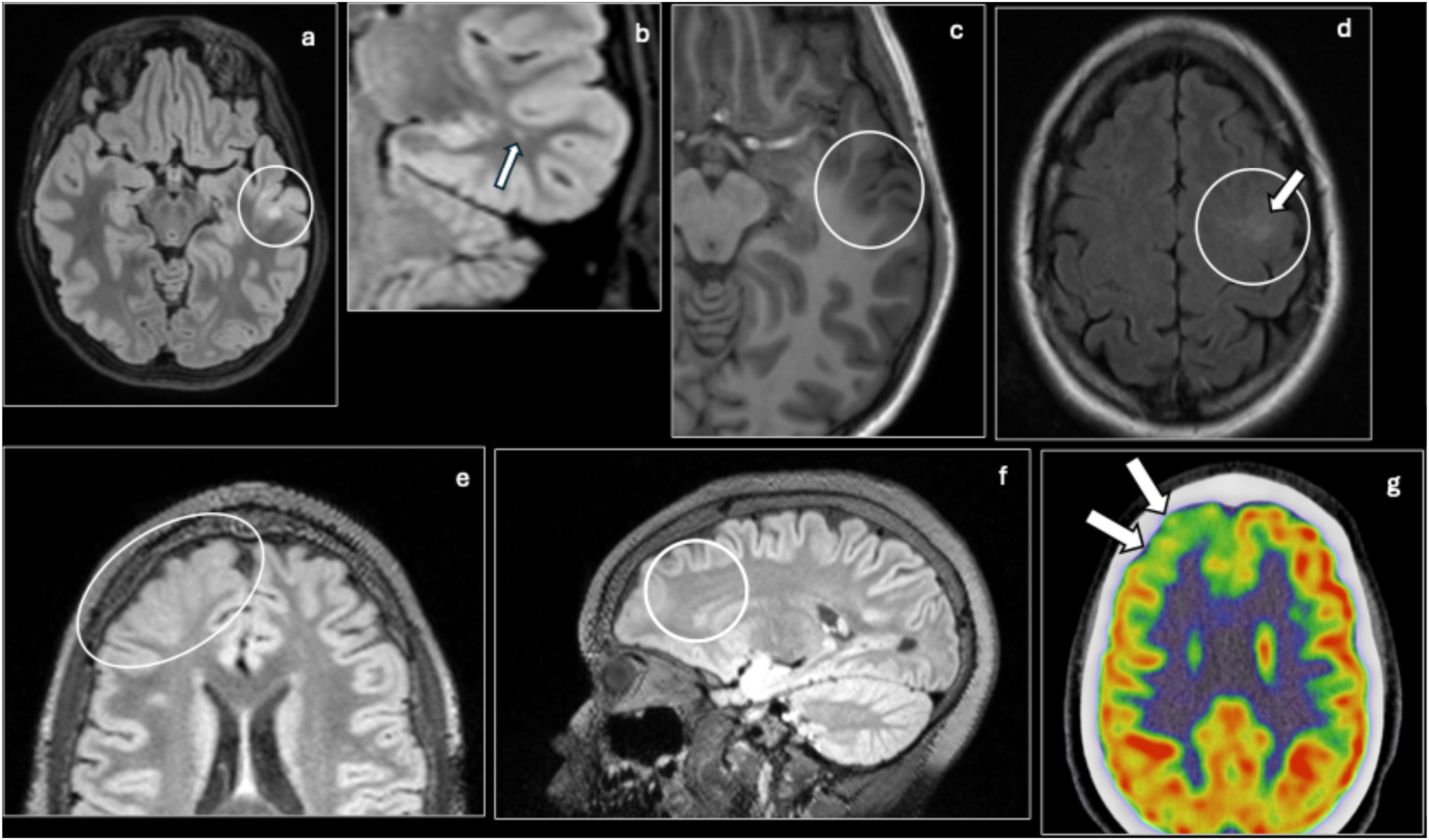
Due to subtle or absent signs, FCD type I is much less commonly detected compared to type II. 18 Compared to isolated FCD type I and type II, FCD type III generally presents with more subtle or absent dysplastic changes in the cortex adjacent to the principal lesion and lacks the transmantle sign seen in type IIb. 20 Diagnosis and identification of epileptogenic foci is improved with careful review of seizure semiology, EEG, and nuclear medicine studies if available. FDG PET is helpful if lesions are not visible or very subtle on MRI and can assist with identification of epileptogenic foci (Figure 2). Furthermore, assessment of the gray–white matter junction may be enhanced by use of FLAWS, which enhances contrast between the gray and white matter, and EDGE, which improves direct visualization of the gray–white matter boundary. 14 In FCD type III, accurate mapping of the entire epileptogenic zone, typically involving the principal lesion and adjacent FCD lesion, with multimodal evaluation is essential to achieving optimal post-surgical seizure control. 21 Invasive EEG may be used in cases with no identifiable epileptogenic foci on imaging. 13 Important alternative diagnoses to consider during evaluation of FCD include tuberous sclerosis, cortical neoplasm (DNET, ganglioglioma, low-grade glioma), incomplete myelination, and gliosis. FCD is distinguished from these other diagnoses by its characteristic focal cortical findings on imaging and lack of systemic clinical features.
Polymicrogyria
Polymicrogyria is a common MCD characterized by small and disorganized gyral convolutions with shallow sulci (“nodular cortex”) resulting from abnormal distribution of cortical neurons. The bilateral perisylvian region is the most commonly affected, but findings may also be unilateral.
13
It is also often associated with other malformative lesions such as cerebellar hypoplasia, callosum agenesis, periventricular nodular heterotopia, and schizencephaly.
9
On MRI of patients with complete myelination (greater than 2 years old), findings include an abnormally thickened and irregular cortex instead of small gyri (Figure 3).
22
In patients 1–2 years old, polymicrogyria presents with an abnormal sulcation pattern. In neonates (or younger than 1 year old), polymicrogyria is best visualized with thin T2WI. Patients 1–2 years old may also be imaged with 3D spoiled-gradient recalled echo (SPGR) T1WI. Alternative diagnoses to consider include microcephaly with simplified gyral pattern, hemimegaloencephaly, pachygyria, and cobblestone malformations. Polymicrogyria can be distinguished from these other diagnoses by its small, irregular gyri without reduced brain volume, hemispheric enlargement, broad and thick gyri, or disorganized cortex with indistinct gray–white matter junction. Polymicrogyria: An 18-month-old with a history of mild global developmental delay, ADHD, buccolingual apraxia and seizures. Axial (a), coronal (b), sagittal right, (c) and sagittal left (d) T1 MR images show thickening and irregularity (ovals) of the perisylvian gray matter bilaterally extending to the perirolandic region, compatible with polymicrogyria.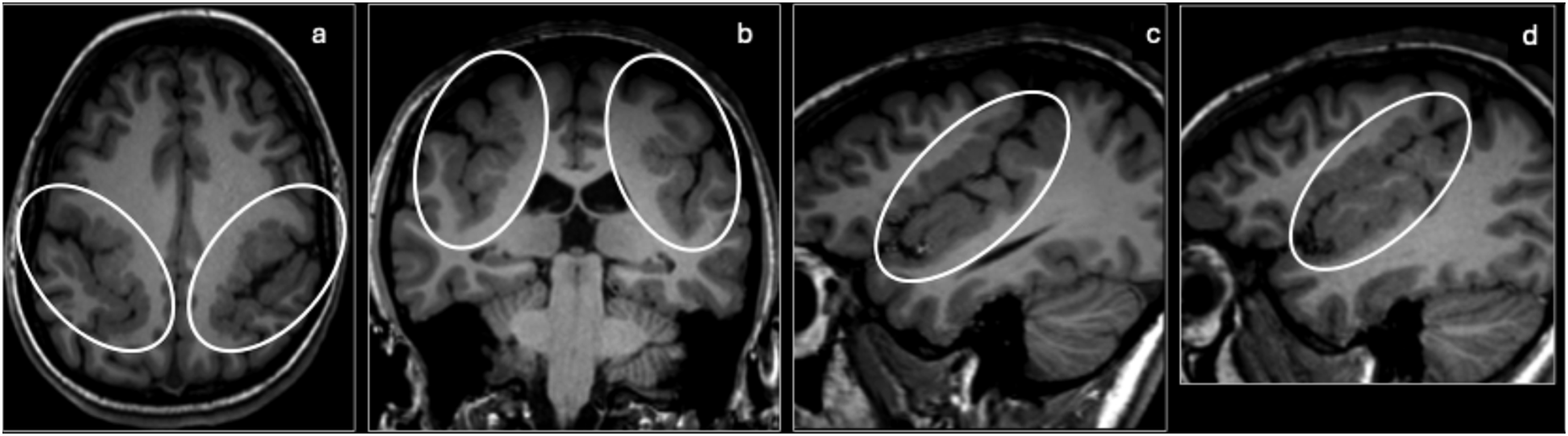
Schizencephaly
Schizencephaly is another cortical malformation characterized by a cleft extending from the cortical surface to the ventricular ependyma.
9
It is a rare condition with a prevalence of 1.54 per 100,000 births.
23
The cleft is lined by dysplastic gray matter, often polymicrogyria, and may occur bilaterally or unilaterally, which is often associated with contralateral polymicrogyria. When the walls of the cleft are separated with CSF present in between, often bilaterally, the malformation is termed open-lipped (Figure 4).
24
When the walls of the cleft are in apposition without CSF in between, usually in unilateral schizencephaly, the type is close-lipped. Multiplanar sequences with or without volumetric acquisition are particularly useful for close-lipped schizencephaly. In patients with incomplete myelination, the gray matter lined cleft is best seen on T2WI.
25
It is important to distinguish schizencephaly from porencephaly cyst, which is an acquired lesion characterized by smooth walls without a gray matter lining or associated cortical malformations, such as polymicrogyria with schizencephaly.
26
Schizencephaly: Axial FLAIR (a), axial T2 (b) show a case of bilateral open-lip (arrows) schizencephaly in a patient with partial agenesis of the corpus callosum.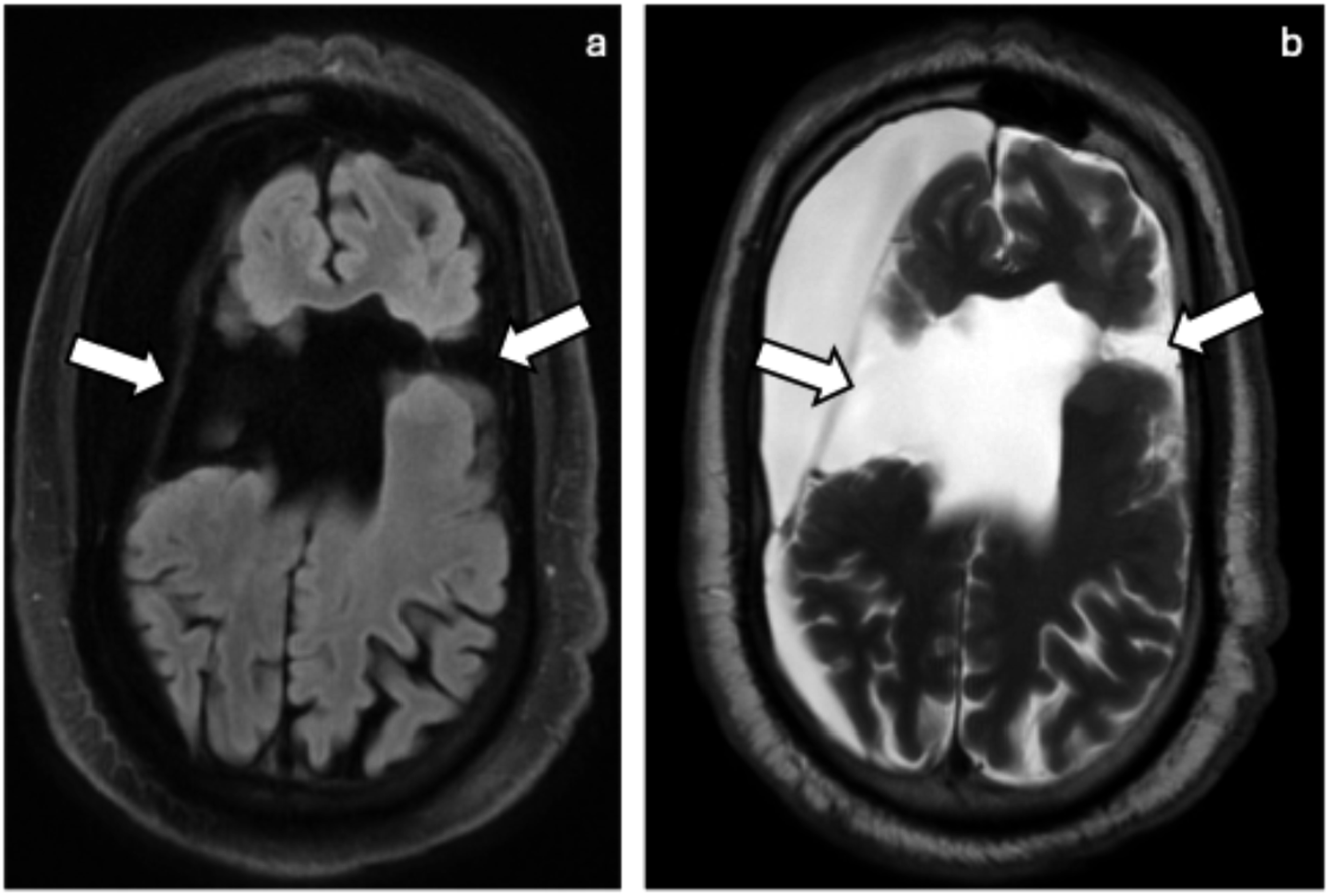
Heterotopic gray matter
Heterotopic gray matter is a rare disorder of abnormal neuronal migration that results in the formation of nodules of ectopic gray matter, consisting of mature neurons and glial cells, without defined lamellar organization.
9
Heterotopia may occur anywhere from the periventricular region to the pia. Periventricular (subependymal) nodular heterotopia is the most common subtype (Figure 5).
27
Subcortical laminar heterotopia (double cortex) is a subtype that involves a continuous or semicontinuous band of gray matter below the cortical mantle.
28
Other subtypes include subcortical nodular heterotopia and pial heterotopias (“cobblestone” cortex). Cobblestone heterotopia is characterized by an irregular, bumpy “cobblestone” brain surface with cortical dysplasia, disruption of the normal cortical contour, and agyria.
29
Helpful MRI sequences for detection of heterotopia include 3D T1WI SPGR in patients with complete myelination and 3D T2WI fast spin echo (FSE). Alternative diagnoses to consider include TSC, closed-lip schizencephaly, ependymal spread of a tumor, polymicrogyria, and congenital CMV. Heterotopic gray matter is distinguished by its non-enhancing, isointense gray matter nodules without mass effect or systemic features. Heterotopic gray matter: Axial T1 Gd+ (a), and axial FLAIR (b) MRI showing incidental heterotopic gray matter (arrows) in a post-operative study.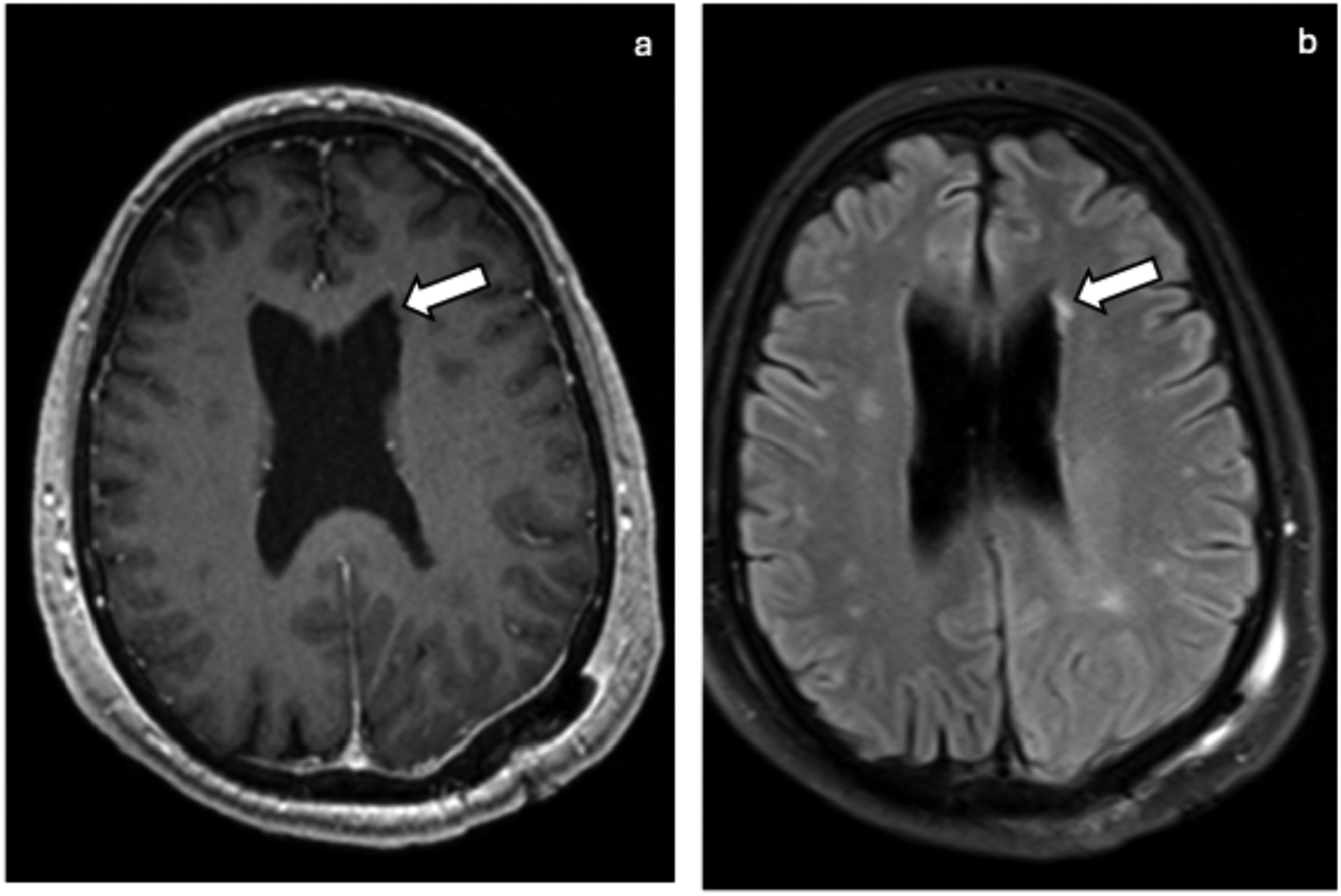
Ulegyria
Ulegyria is a condition that usually results from perinatal global hypoxic ischemic brain injury producing cortical and subcortical lesions such as shrunken and flattened cortex and multiple cerebral convolutions (Figure 6).
30
In children with a history of term hypoxic-ischemic injury and cortical injury on imaging, ulegyria was identified in 66% of patients.
31
Ulegyria is most commonly centered around the deepest portion of a gyrus and usually in the parasagittal watershed and posterior cerebral artery distributions.
32
On MRI, ulegyria is characterized by a unique mushroom-shaped appearance resulting from small atrophic circumvolutions at the bottom of a sulcus underlying an intact gyral apex. Findings may be unilateral or bilateral. Associated clinical findings include cerebral palsy, mental disability, and intractable focal seizures often with visual auras and mean onset at 4 years old.
13
Ulegyria: Axial FLAIR (a) and T1 Gd+ (b) show a shrunken and flattened gyrus (ovals) in the left parietal lobe compatible with ulegyria.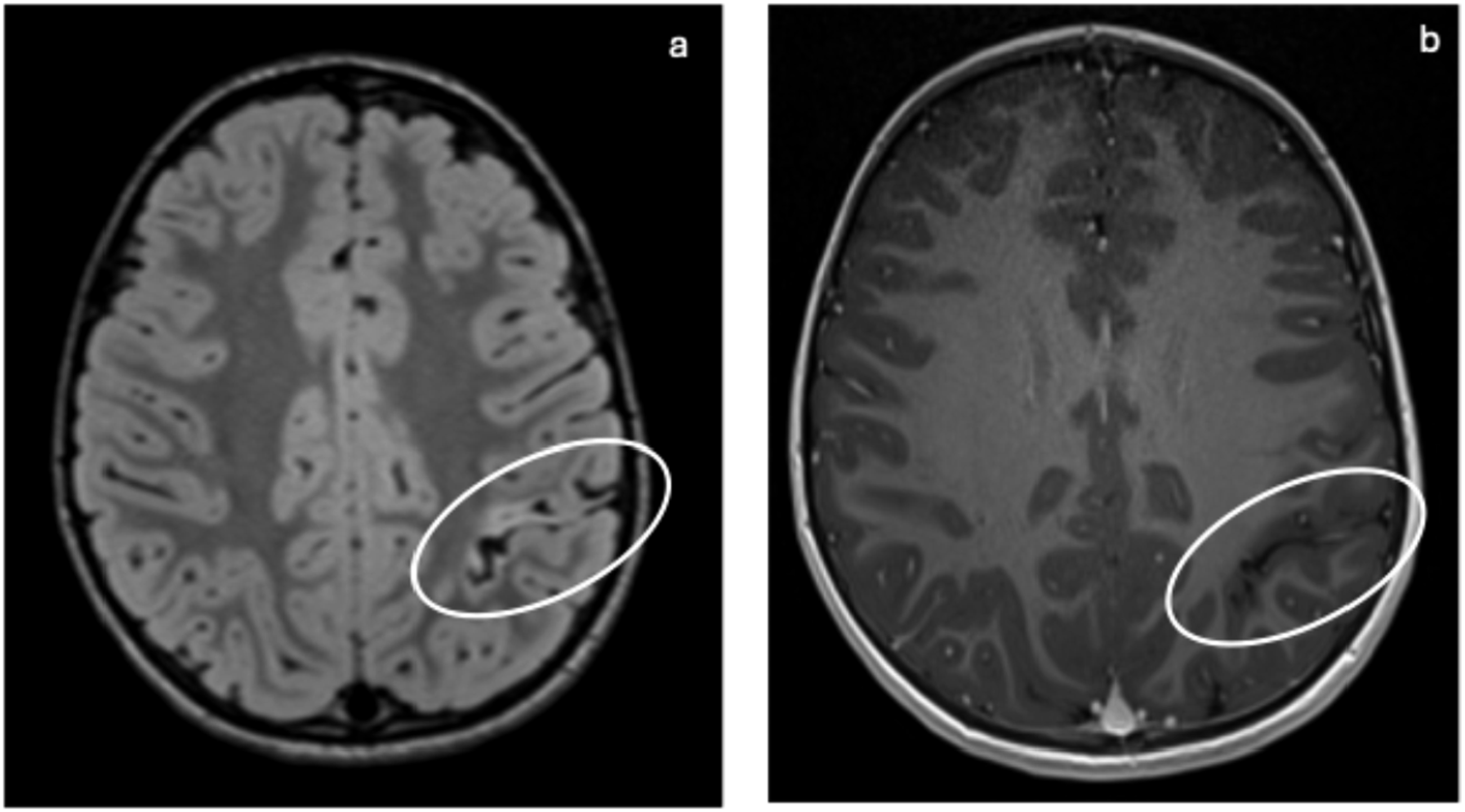
Ectopic posterior pituitary
Ectopic posterior pituitary (EPP) is a rare condition characterized by the absence, truncation, or thread-like appearance of the pituitary stalk leading to positioning of the posterior pituitary gland outside of the sella turcica. EPP is thought to arise from defective neuronal migration during embryogenesis.
33
EPP is best seen on midline sagittal T1WI MRI and is commonly located along the median eminence of the tuber cinereum or truncated pituitary stalk (Figure 7).
34
As age increases, the pituitary bright spot dims and may appear absent on T1WI.
35
Commonly associated abnormalities include heterotopia, optic nerve hypoplasia, and corpus callosum abnormalities. Alternative diagnoses to consider include surgical or traumatic stalk transection, central diabetes insipidus, and hypothalamic lipoma (in the tuber cinereum). EPP can be distinguished from these diagnoses by gradual progression of panhypopituitarism, presence of ectopic bright spot seen on fat-specific imaging. Ectopic posterior pituitary: Sagittal T1 Gd+ (a), sagittal FLAIR (b), and coronal T1 w/o contrast (c) MRI show an intrinsically T1 hyperintense nodule (arrows) in the superior aspect of the pituitary infundibulum which enhances in the early dynamic phase and remains hyperintense on Gd + FS. The pituitary T1 posterior bright spot (arrow) is missing. Findings are suggestive of an ectopic posterior pituitary.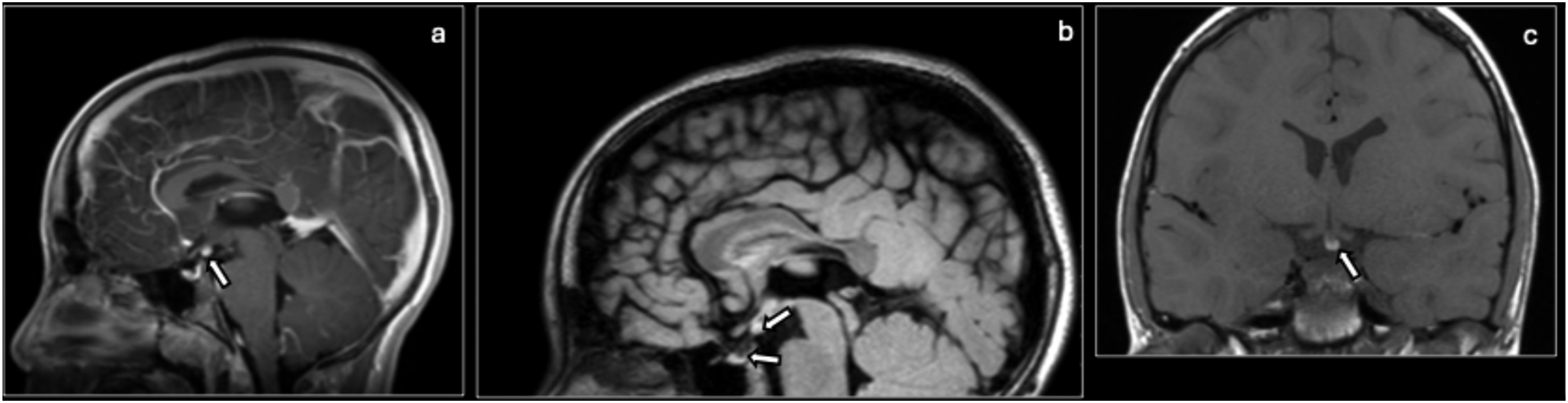
Septo-optic dysplasia
Septo-optic dysplasia (SOD) is a rare congenital neurodevelopmental disorder with a wide range of clinical presentations ranging from normal cognitive and neurological function to developmental delay and epilepsy. SOD has prevalence at birth of 1.41 per 10,000 live births.
36
It defined by the presence of at least 2 of the following three features: optic nerve hypoplasia (often asymmetric), midline brain abnormalities (typically malformations in the septum pellucidum and corpus callosum), and pituitary gland dysfunction (often ectopic posterior pituitary).
37
There may be flat-roofed ventricles and inferiorly pointed frontal horns (Figure 8).
38
The presence of additional cerebral cortical malformations such as polymicrogyria and schizencephaly is known as SOD-plus syndrome. Imaging of SOD involves MRI with 3D acquisitions or multiplanar reformation (MPR) such as 3D GRE-based T1 and 3D-balanced steady state (SS) GRE. Septo-optic dysplasia: Coronal T1 (a), coronal T2 (b) MR show a hypoplastic left optic nerve (arrow) with increased T2 signal (arrow). Coronal T2 (c) shows an absent septum pellucidum (arrow) and flat-roofed lateral ventricles (arrow).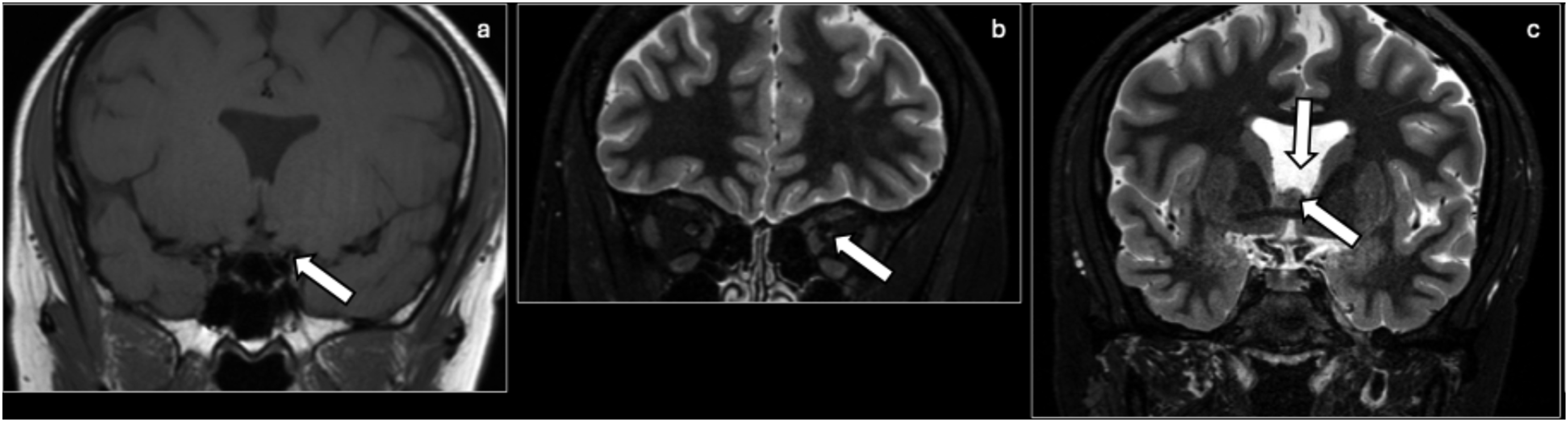
Cephalocele
A cephalocele is another congenital anomaly that is characterized by extracranial extension of intracranial contents through a defect in the skull. These contents can include meninges, brain tissue, and CSF. It occurs in 0.8–4 per 10,000 live births.
39
Cephalocele more commonly occurs in the occipital region but can also occur at the frontoethmoidal, parietal (atretic or cranial vault), or basal (nasopharyngeal) regions.
40
On MRI imaging, a cephalocele will have a heterogeneous signal intensity of contents reflecting the brain parenchyma and CSF (Figure 9). MRV can help to characterize venous relationships to the cephalocele, particularly in the occipital region. Alternative diagnoses to consider include dermoid or epidermoid cyst, nasal glial heterotopia (“nasal glioma”), sinus pericranii, and vascular anomalies such as neoplasms and malformations. These alternative diagnoses lack direction connection to the intracranial compartment and a skull defect or will show venous communication, such as in the case of sinus pericranii. Cephalocele: Coronal FLAIR (a, b), and axial T2 (c) of the brain showing a right-sided temporal lobe encephalocele (arrows).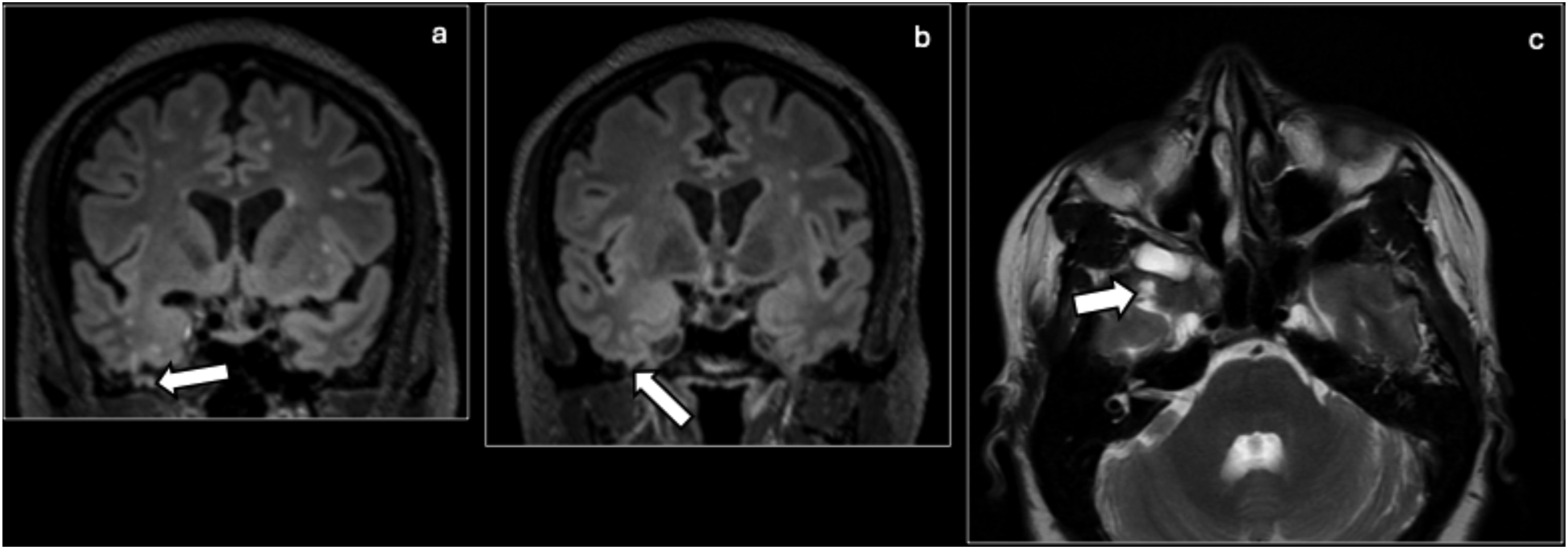
Tuberous sclerosis complex
Tuberous sclerosis complex (TSC) is a rare neurocutaneous disorder characterized by hamartomatous lesions or “tubers” in various organs and systems.9,13 The incidence is estimated to be between 1:6,000 and 1:10,000 live births.
41
TSC often initially manifests as infantile spasms in the first months of life, progressing to intellectual disability and intractable focal seizures. In the CNS, TSC is characterized by cortical tubers, subependymal nodules, subependymal giant cell astrocytoma, and white matter lesions. On MRI imaging, tubers change with myelination, appearing hyperintense on T1 and hypointense on T2 in neonates and appearing hypointense on T1 and hyperintense on T2 with poorly defined borders in older children (Figure 10).9,42 Cortical tubers may be difficult to differentiate between FCD. Unlike FCD, cortical tubers tend to be multifocal, affect any part of the cortex, are often associated with calcifications, and are present with other features of TSC. It is also important to consider cavernomas as a differential diagnosis in the evaluation of calcified tubers. Other alternative diagnoses include gray matter heterotopia, cortical dysplasias, and germinal matrix. TSC can be distinguished by the presence of subependymal nodules and systemic features. Tuberous sclerosis: Axial FLAIR (a-c) and axial SWI (d) show scattered foci of FLAIR hyperintensities (circle) in multiple lobes, some of which show blooming artifact (circle) on SWI, compatible with calcified cortical tubers in the setting of TSC. Subependymal nodules (arrow) and multiple scattered linear or radial bands of FLAIR hyperintensities (arrow) are also shown. Axial FLAIR (e), coronal FLAIR (f), and coronal T2 at multiple planes (g–i) MRI show multiple subtle juxtacortical (circles)/cortical T2/FLAIR hyperintensities (arrows) predominantly in the bilateral frontal lobes, which may represent focal cortical dysplasia versus cortical tubers.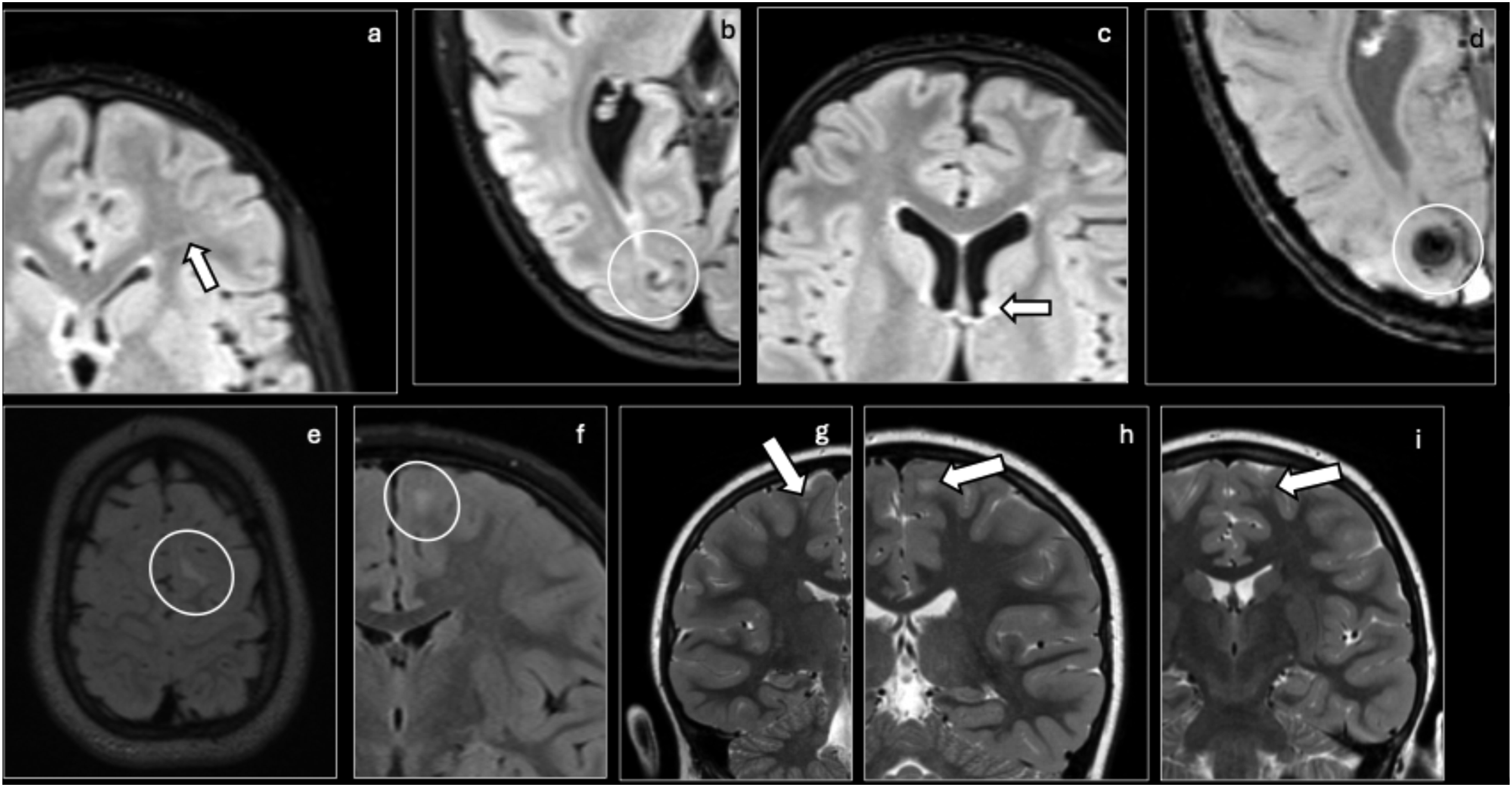
Sturge–Weber syndrome
Sturge–Weber syndrome (SWS) is another rare congenital neurocutaneous disorder characterized by abnormal cortical venous development with an incidence of 3.08 per 100,000 people per year.43–45 Clinically, patients with SWS often present with focal antiepileptic-resistant seizures, hemiparesis, cognitive impairment, and headaches. The hallmark features of SWS include facial capillary malformation, known as a port-wine stain typically involving the first division of the trigeminal nerve, leptomeningeal vascular malformations, and ocular abnormalities such as glaucoma. Imaging findings include ipsilateral leptomeningeal enhancement, ipsilateral enlarged choroid plexus, cortical atrophy, cortical calcification with a tram track appearance, calvarial thickening, and sinus hyperpneumatization (Figure 11).
44
Alternative diagnoses to consider include an acquired meningeal process, PHACES syndrome, blue rubber bleb nevus syndrome, Klippel–Trenaunay syndrome, and Wyburn–Mason syndrome. SWS is distinguished from these diagnoses by the presence of a port-wine stain with ipsilateral leptomeningeal enhancement and ocular involvement. Sturge–Weber syndrome: Coronal T2 (a), axial T1 (b), and axial FLAIR (c) showing cortical atrophy (circle, arrows) associated with increased signal on FLAIR of the left cerebral hemisphere (arrow), most pronounced in the left frontal lobe, compatible with the patient’s known Sturge–Weber syndrome.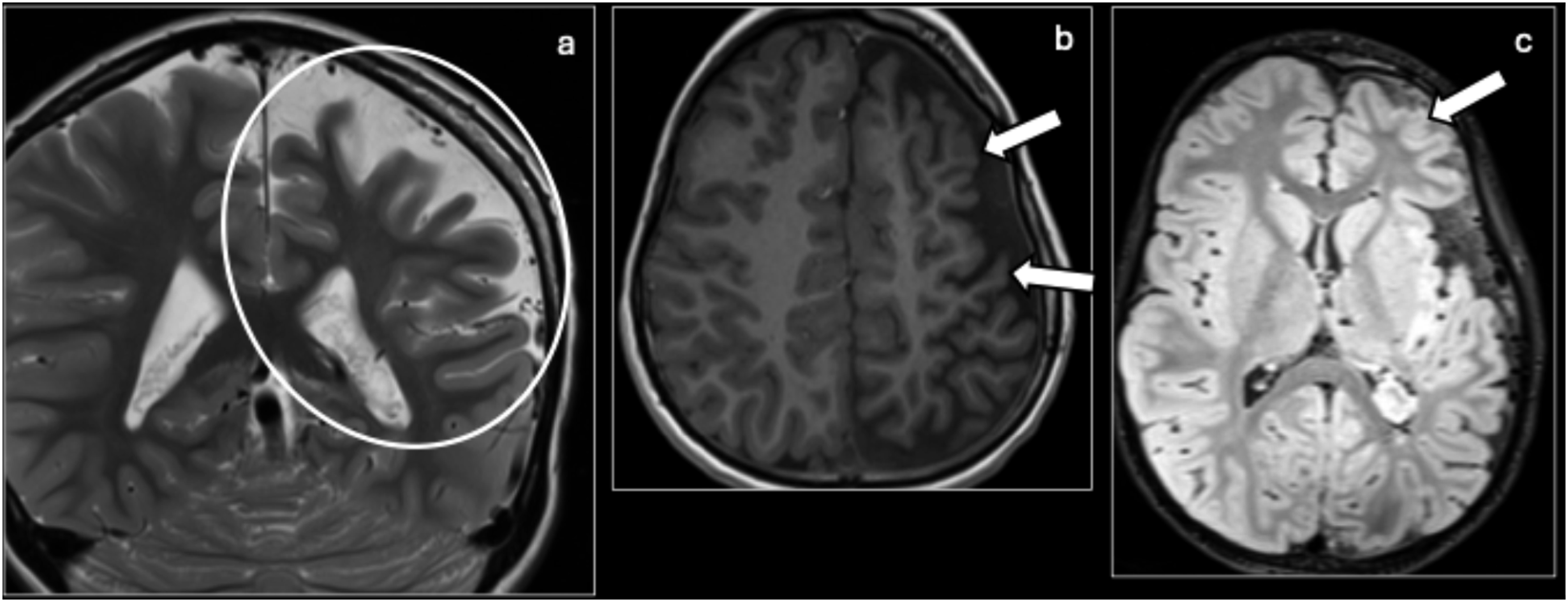
Leigh disease
Leigh disease or subacute necrotizing encephalomyelopathy is a rare neurometabolic disorder characterized by progressive neurodegeneration typically manifesting in infancy or early childhood.
46
The prevalence is estimated to be between 1:30,000 and 1:40,000.
47
Clinically, Leigh disease presents with developmental delay, hypotonia, seizures, ataxia, and respiratory. Lactate may be elevated in the blood, CSF, as well as within the brain (on MR spectroscopy) due to mitochondrial dysfunction. The hallmark feature of Leigh disease is symmetrical “speckled” lesions in the basal ganglia or brainstem on MRI, which appear as hyperintensities on T2 and hypointensities on FLAIR (Figure 12).
48
Alternative diagnoses to consider include hypoxic ischemic encephalopathy, Wilson disease, near drowning, kernicterus, Huntington disease, and encephalitis. Leigh disease is distinguished from these conditions by its progressive neurological symptoms, symmetric lesions in the basal ganglia on MRI, and elevated lactate on various testing modalities. Leigh disease: Axial FLAIR (a), T2 (b), and SWI (c) showing increased signal on T2, decreased signal on FLAIR, and susceptibility on SWI (ovals) in the bilateral putamen, compatible with the history of Leigh syndrome in this patient.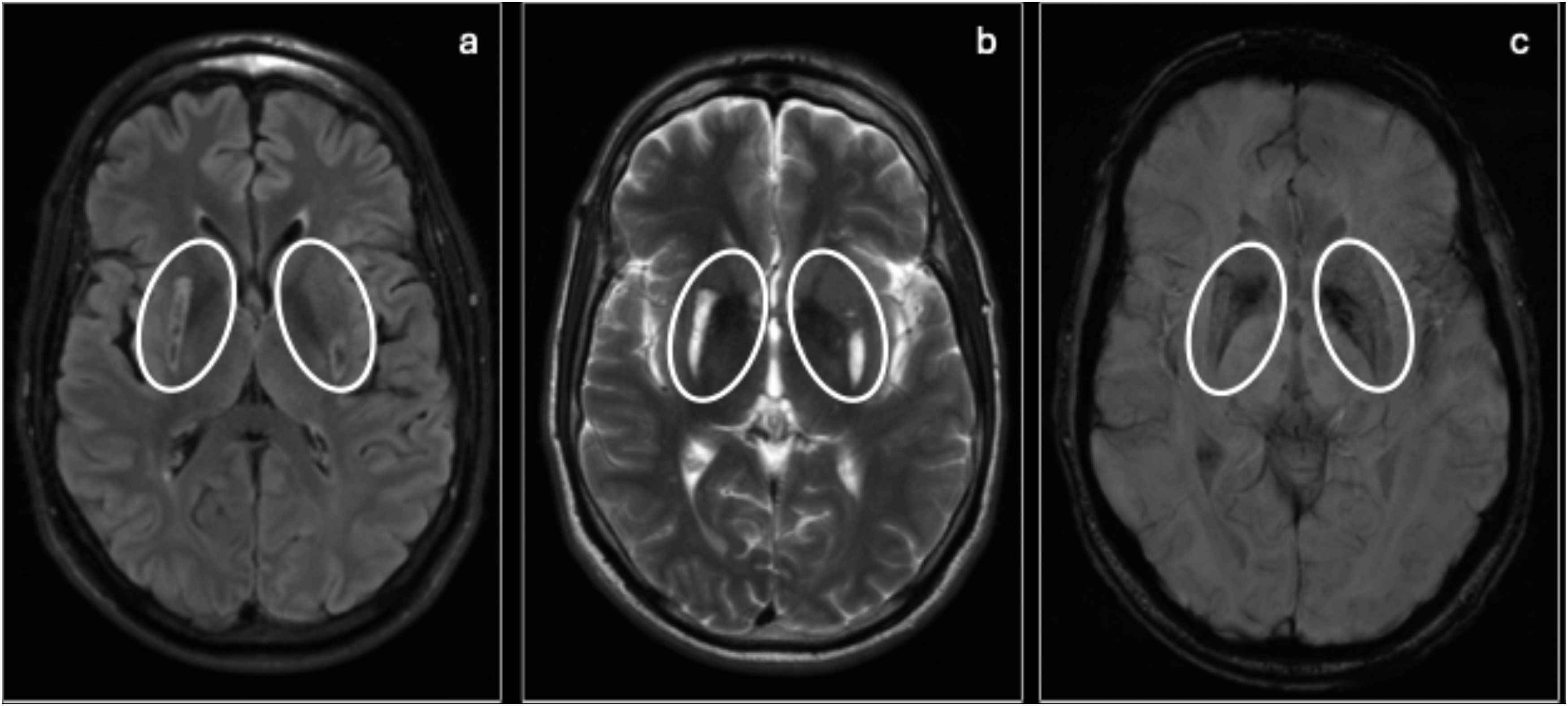
Inflammatory/infectious etiologies
Rasmussen encephalitis
Rasmussen encephalitis is a rare immune-mediated progressive hemispheric encephalopathy characterized by childhood onset, intractable epilepsy, hemiparesis, and neurologic decline.
9
The incidence is estimated to be 2.4 per 10 million people aged less than 18 per year.
49
On MRI, there is progressive atrophy of one cerebral hemisphere, usually beginning in the insula, perisylvian region, caudate head, and putamen, with cortical hyperintensities on T2 and FLAIR (Figure 13).
13
Serial imaging may show fluctuations with regression followed by reappearance of lesions on FLAIR in the cortical and subcortical regions, predominantly in the frontoinsular and parietal regions.
50
There may also be a crossed cerebellar diaschisis. Alternative diagnoses to consider include SWS, other autoimmune encephalitis (often with bilateral findings), and Dyke–Davidoff–Masson syndrome. Rasmussen encephalitis is distinguished by its progressive, unilateral cerebral atrophy with T2/FLAIR hyperintensities and without enhancement. Rasmussen encephalitis: Axial T1 (a), coronal T2 (b), and coronal FLAIR (c) MRI of the brain showing left cerebral hemisphere volume loss (arrows, circles) compared to the right, most severe in the parietal lobe, and least severe in the frontal lobe. The caudate head and basal ganglia appear unremarkable.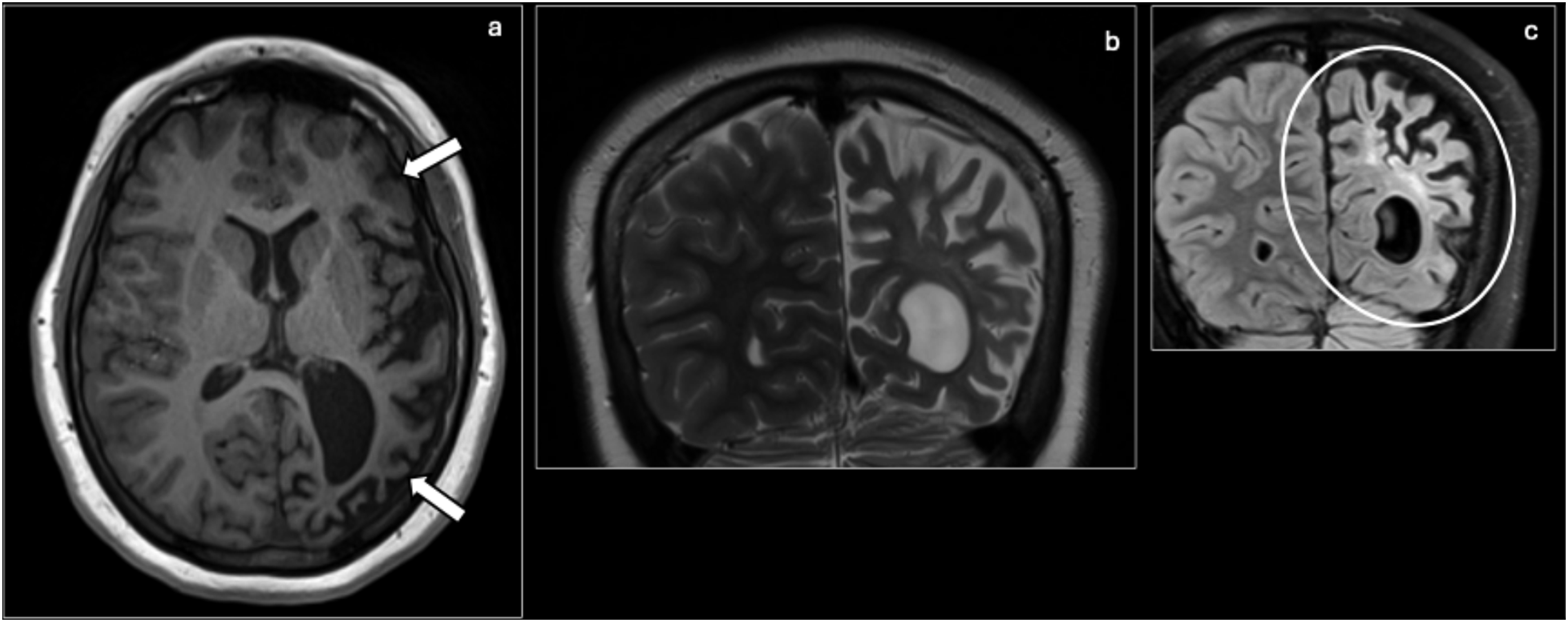
Meningoencephalitis
Meningoencephalitis is a condition of simultaneous inflammation of the meninges and brain parenchyma caused by a wide variety of infectious and non-infectious agents.
51
It is typically a clinical diagnosis that presents with fever, neck stiffness, altered mental status, seizures, and focal neurological deficits with confirmation by CSF analysis showing pleocytosis, elevated protein, and elevated inflammatory markers. Imaging plays a role in identifying etiologies and assessing complications such as empyema, ischemia, hydrocephalus, cerebritis, and ventriculitis. Location of abnormalities provides a clue for the etiology with cerebral convexities suggesting a bacterial agent; basilar meninges suggesting TB, neurosyphilis, cryptococcus, sarcoidosis, and lymphoma; and meningoencephalitis suggesting a viral agent.
52
These lesions may appear as hyperintense signals in sulci, cisterns, or brain parenchyma on T2 and FLAIR with enhancement of the meninges (Figure 14). It is important to note that meningitis may be present even with normal imaging. Meningoencephalitis: Coronal T1 Gd+ (a), coronal T2 (b), and axial T1 Gd+ (c) showing frontotemporal and subtle cortical enhancement (arrows, circle) with associated increased T2 signal (circles) concerning for meningoencephalitis.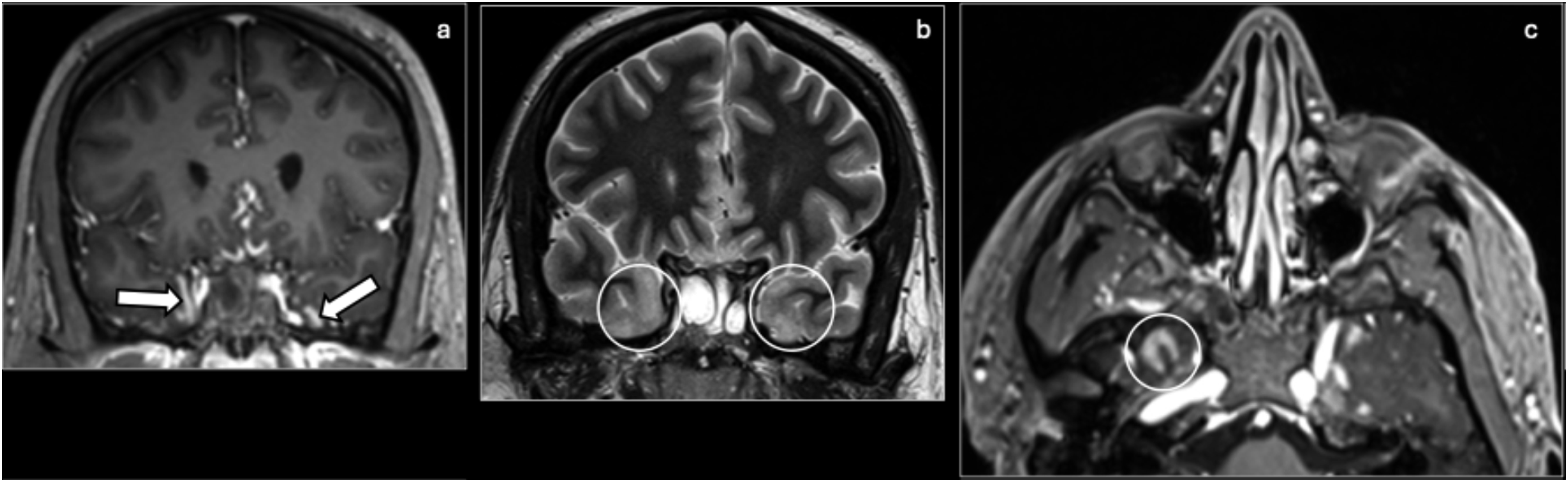
Autoimmune encephalitis
Autoimmune encephalitis (AE) is an immune-mediated disease with antibody-mediated inflammation of the brain. It has an incidence of 0.8 per 100,000 person-years.
53
Paraneoplastic AE may occur as a neurologic complication of underlying malignancy outside the CNS, most commonly small cell lung carcinoma.
54
The most common clinical paraneoplastic syndrome is limbic encephalitis. Clinically, limbic encephalitis presents with rapid onset of psychiatric symptoms, cognitive impairment, temporal lobe seizures, and movement disorders. On imaging, limbic encephalitis is characterized by hyperintensities in the mesial temporal lobes and limbic system (Figure 15).
55
It is important to note that brain MRI is positive in only 30–50% of patients with anti-NMDAR paraneoplastic encephalitis.
56
Alternative diagnoses to consider include HSV encephalitis, diffuse astrocytoma, status epilepticus, and gliomatosis cerebri. Limbic encephalitis, which may mimic HSV encephalitis, is typically associated with a subacute or chronic course. Paraneoplastic encephalitis: Axial T2 pelvic MR (a) from a patient presenting with multiple episodes of seizures. The patient underwent several brain MRI scans which had failed to reveal an etiology for the seizures. A full-body MRI subsequently revealed an ovarian teratoma (arrow). The seizures were then found to be related to anti-NMDAR encephalitis. Axial FLAIR MR (b) from a different patient shows abnormal hyperintensity (circles) in the bilateral medial temporal lobes, characteristic of limbic encephalitis, the most common paraneoplastic syndrome.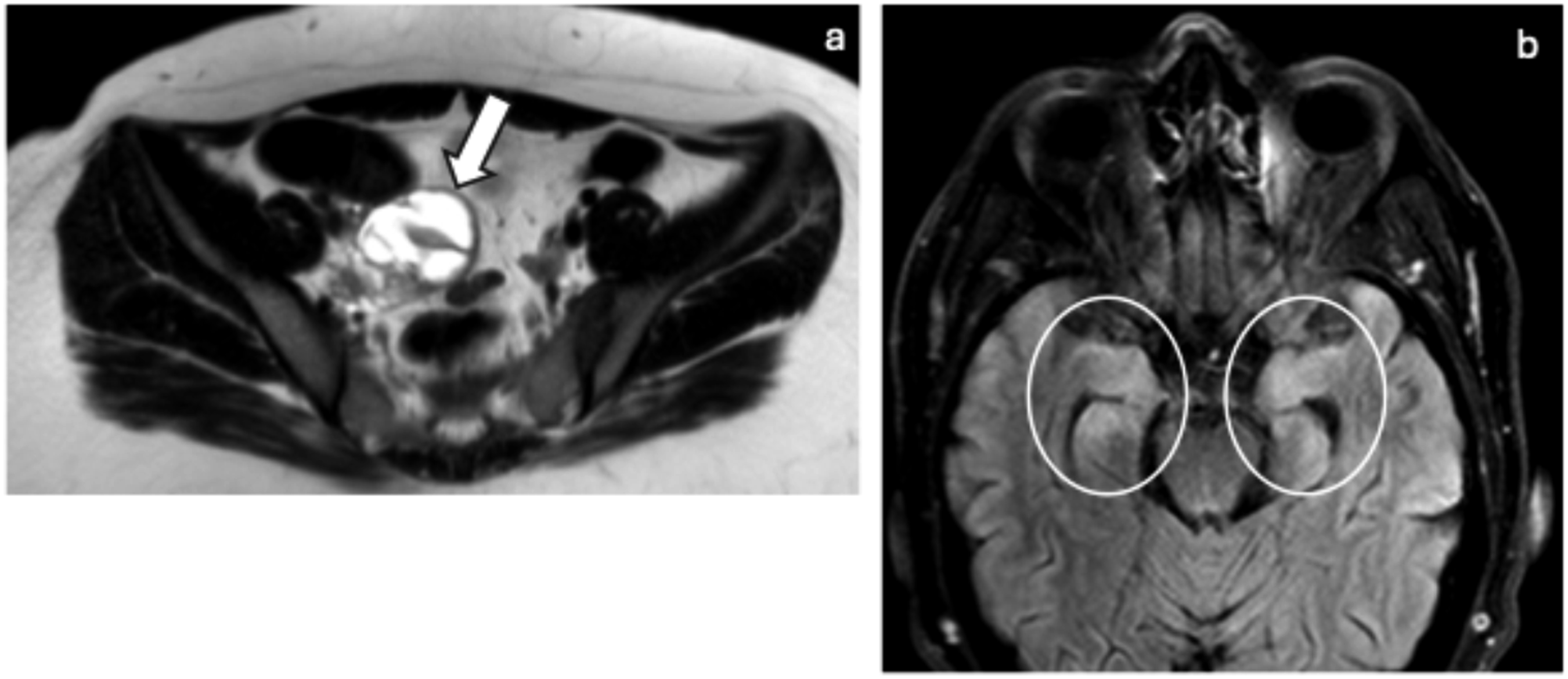
Neoplastic etiologies
Long-term epilepsy associated tumors
Long-term epilepsy associated tumors (LEATs) are encountered in approximately 30% of patients with long-term pharmaco-resistant epilepsy.
13
The most common of these are gangliogliomas, which appear as cortically based enhancing cystic nodules on imaging (Figure 16).
57
They are most frequently found in the temporal lobe and can be supratentorial but can also be infratentorial. Calcifications are found in about 50% of cases. Most cases are also associated with cortical disorganization (FCD type IIIb). Differentiating features of FCD include the presence of cortical thickening and transmantle sign. Other alternative diagnoses to consider include pilocytic astrocytoma, DNET, xanthoastrocytoma, diffuse astrocytoma, and oligodendroglioma. Ganglioglioma: A 19-year-old female presenting due to “flutter episodes” for approximately a year and one generalized tonic-clonic seizure. Coronal T2 (a), axial FLAIR (b), and axial post-contrast (c) MR images show an approximately 1 cm rounded enhancing intra-axial lesion (circles) in the lateral aspect of the right superior frontal gyrus with no surrounding edema or mass effect. The patient underwent resection of the lesion, and surgical pathology confirmed the diagnosis of ganglioglioma.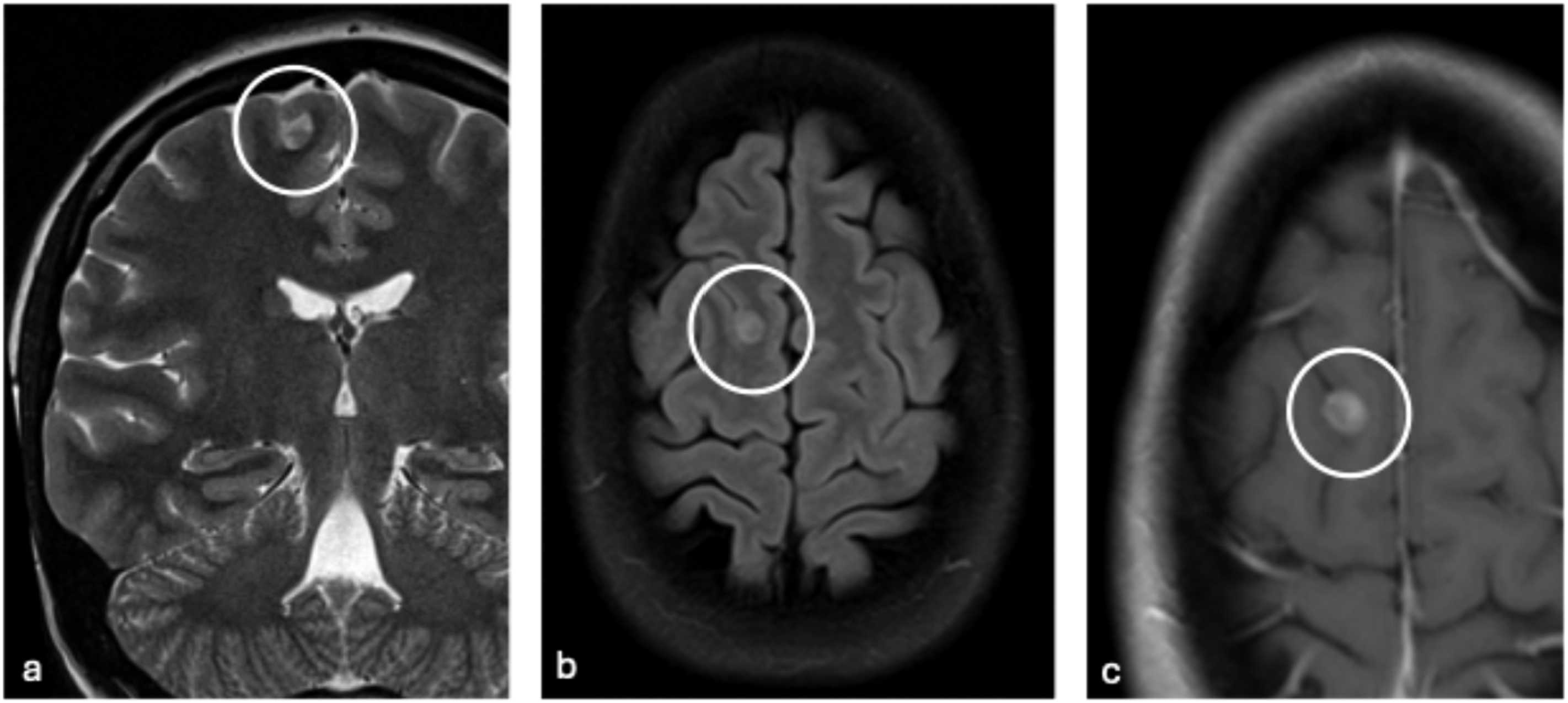
Dysembryoplastic neuroepithelial tumor
Dysembryoplastic neuroepithelial tumor (DNET) is a benign, slow growing, mixed-glial neuronal neoplasm.
13
On imaging, DNETs appear as a T2-hyperintense “bubbly” cortical mass with minimal or no mass effect, no peritumoral edema, and typically with sharp demarcation (Figure 17).
58
Enhancement is significantly less common than with gangliogliomas. Some DNETs with less distinct margins may have associated focal cortical dysplasia. The most common site is the medial temporal lobe. Alternative diagnoses to consider include ganglioglioma, FCD, neuroepithelial cyst, pleomorphic xanthoastrocytoma, and angiocentric glioma. Dysembryoplastic neuroepithelial tumor: A 46-year-old male with new-onset seizures. Coronal T1 (a), T2 (b), FLAIR (c), and axial T2 (d) show a multilobar, T1 hypointense, T2/FLAIR hyperintense, “bubbly” lesion (ovals) in the right temporal lobe. The lesion was resected and pathology was consistent with dysembryoplastic neuroepithelial tumor (DNET).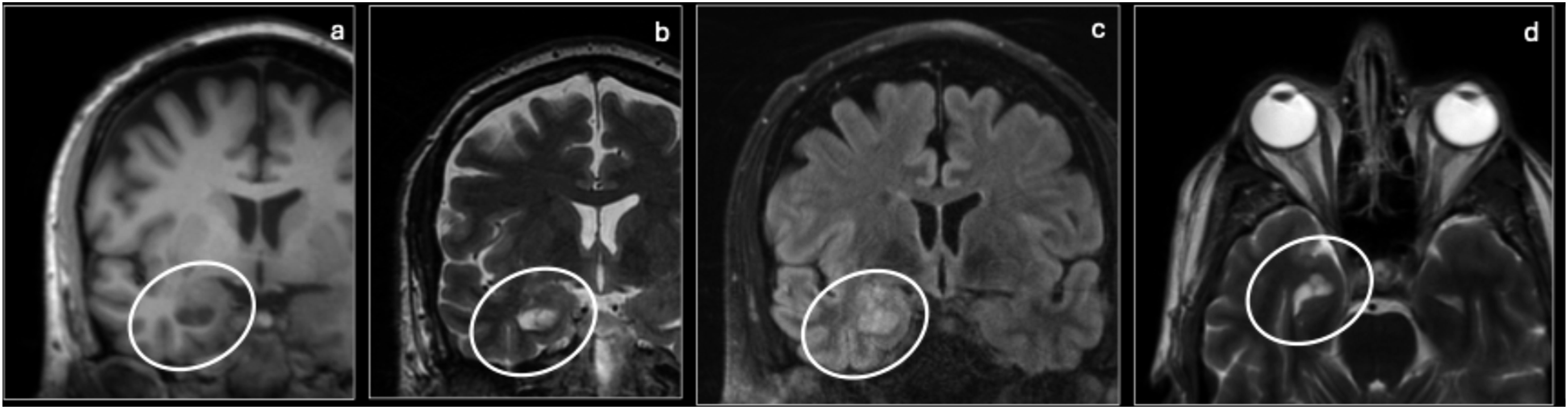
Multinodular and vacuolating nodular tumor
Multinodular and vacuolating nodular tumor (MVNT) is a rare, benign neuronal tumor characterized by multiple nodules and vacuolated neuronal cells, primarily encountered as an epileptogenic lesion in the subcortical white matter.
59
MVNT appears as a cluster of variably sized, confluent, and discrete nodules, often in a U-shaped configuration with no mass effect (Figure 18). They can be found in the deep cortical ribbon and superficial white matter sparing the superficial gray matter and can also be found in the vermis of the cerebellum. On MRI, the lesions are isointense with the cortex on T1WI, hyperintense on T2WI/FLAIR, very bright on DWI, and exhibit no enhancement. Alternative diagnoses to consider include DNETs, FCD, and enlarged perivascular spaces. Multinodular and vacuolating neuronal tumor: Coronal T2 (a) and FLAIR (b) MRI show confluent and multiple discrete T2/FLAIR hyperintense nodules (ovals) along the undersurface of the cortex within the left inferior parietal lobe. Please note the U-shape configuration of the clusters (arrows) and the lack of mass effect in the magnified coronal T2 (c). Coronal pre- (d) and post-Gd+ (e) T1 MRI show the isointense nodules (circles) compared to the cortex.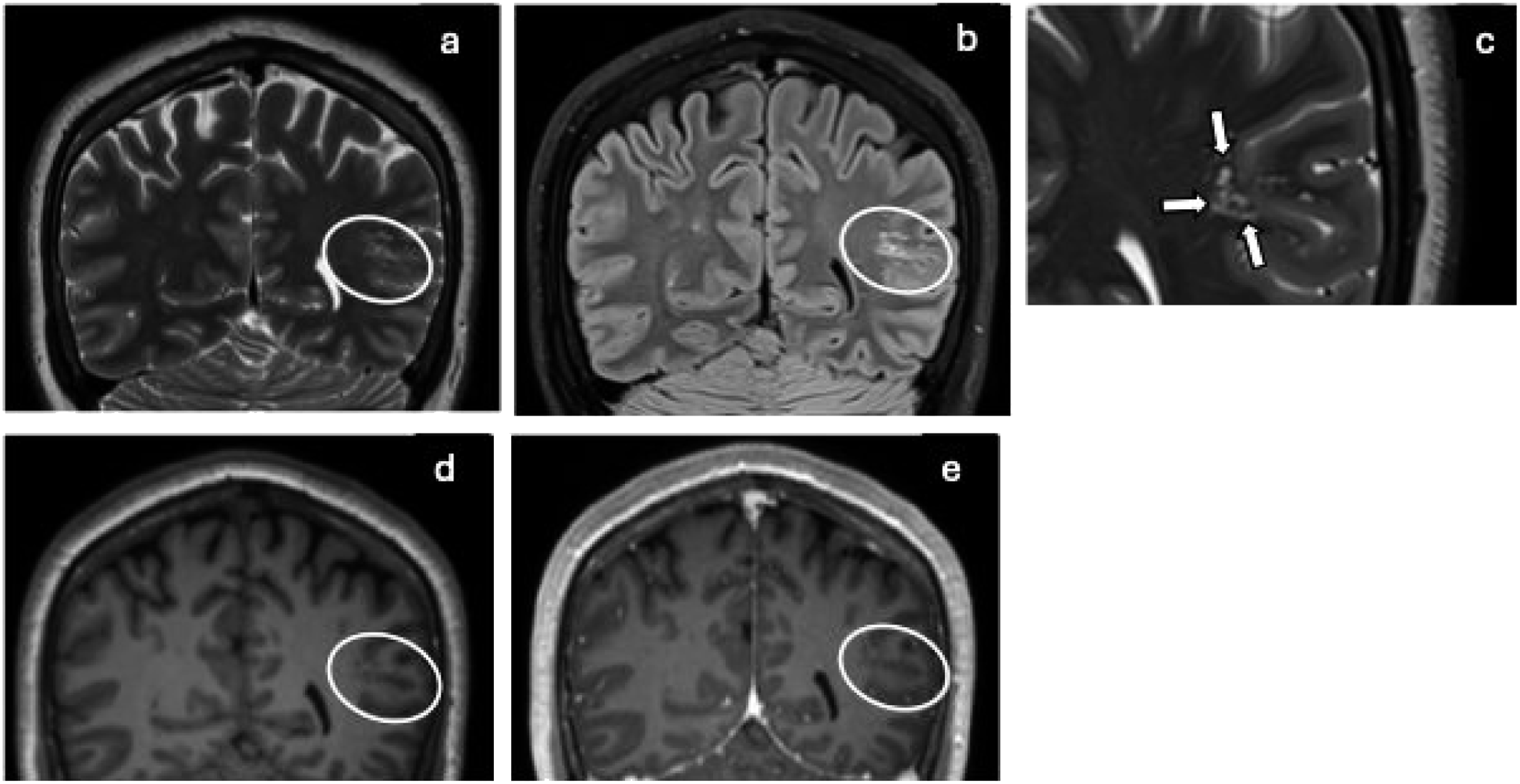
Miscellaneous etiologies
Cerebral cavernous malformations
Cerebral cavernous malformations, also known as cavernomas, are benign vascular lesions made up of dilated sinusoids lined by thin, immature walls containing blood products of various ages.
9
This results in hamartomatous or “berry-like” of vascular spaces without any intervening brain parenchyma.
60
The peak incidence for cavernomas is 4–60 years old, and they are estimated to occur in 0.5% of the general population.
61
On imaging, cavernomas have a heterogenous or “popcorn” appearance on T2, a hypointense hemosiderin rim, hypointensity on SWI, and absent or minimal enhancement (except in cavernomas with associated developmental venous anomaly) (Figure 19).
62
Sizes may range from microscopic to giant (larger than 6 cm), and they are most common in the supratentorial region compared to the infratentorial region and spinal cord. Alternative diagnoses to consider include diffuse axonal injury, intracerebral hematoma, neoplasm, and capillary telangiectasia. Cavernomas are distinguished by their “popcorn” appearance, hemosiderin rim, and lack of significant enhancement and edema. Cavernous malformation: Axial T2 (a), T1 (b), FLAIR (c), and SWI (d) MRI of the brain showing heterogenous signal (ovals) with a hypointense rim on T2, T1, and FLAIR of the right uncus as well as a decreased signal on SWI.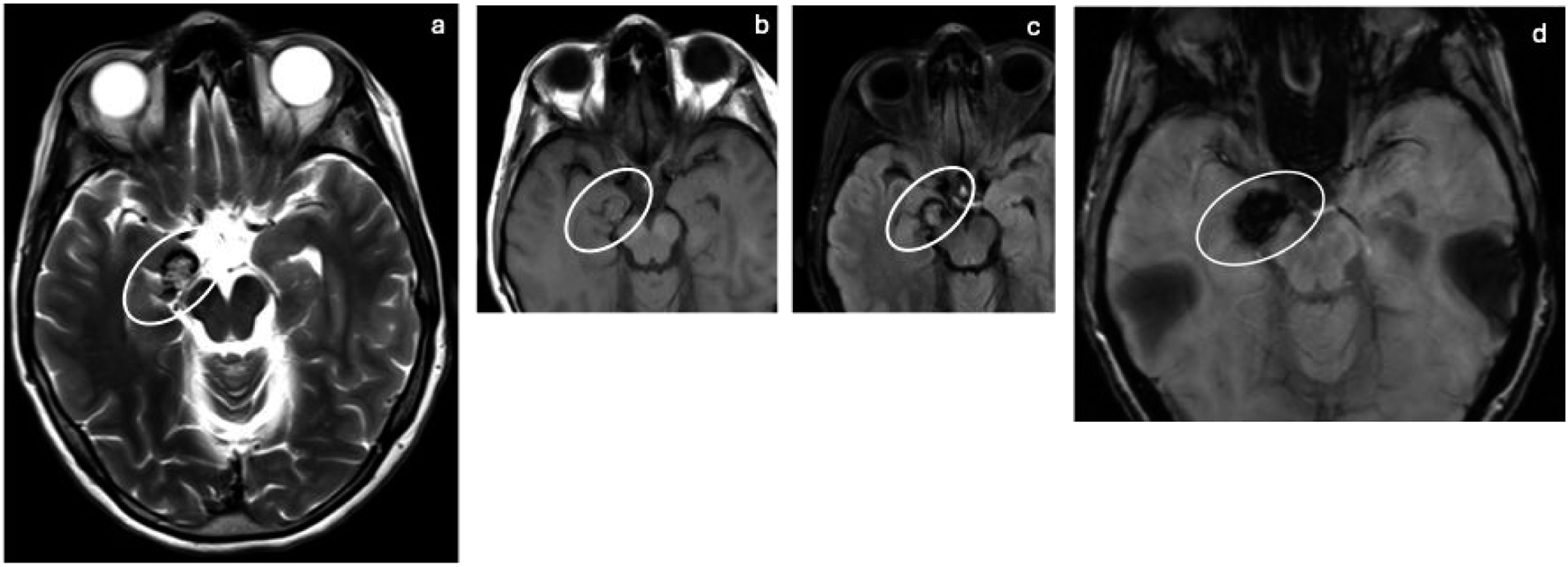
Gliosis
Gliosis is a relatively common reactive process occurring after some time following most types of CNS injuries, such as trauma, infection, ischemia, and neurodegenerative disease.
1
The prevalence of gliosis varies based on the underlying condition. In patients undergoing epilepsy surgery, glial scars were found in 4.9% of patients.
15
Gliosis is characterized by focal proliferation and activation of glial cells, particularly astrocytes. Gliosis can be asymptomatic or be a seizure-focus. The presenting features of gliosis are typically related to the underlying condition causing the gliosis. On imaging, gliosis produces no volume loss, no mass effect, and appears hyperintense on T2/FLAIR and mildly hypointense on T1, with mildly facilitated diffusion on ADC (Figure 20).
63
It may also mimic a low-grade astrocytoma. On CT, small or subtle areas of gliosis may be invisible but more pronounced gliosis produces white matter hypoattenuation. Gliosis: Axial T1 (a), T2 (b), and FLAIR (c) MRI of the brain showing decreased signal on T1 and increased signal on T2 and FLAIR associated with bifrontal gliosis (arrows).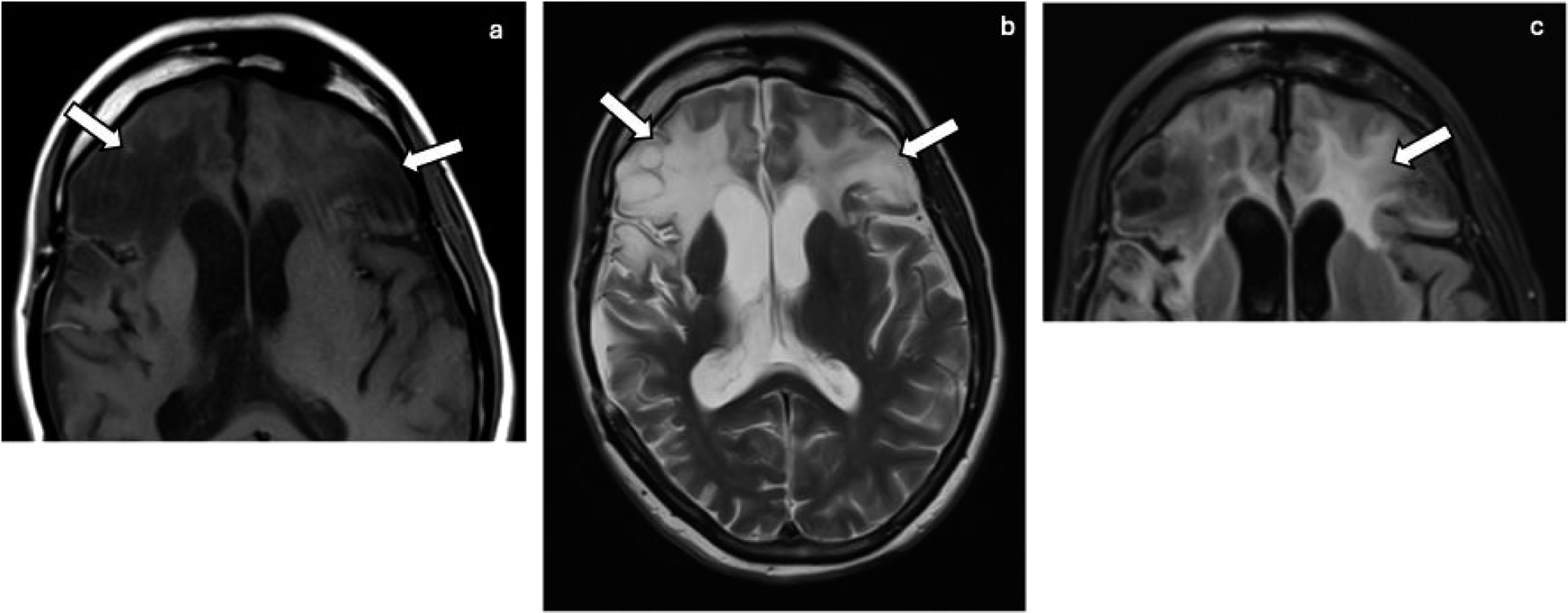
Dual pathology
Dual pathology refers to the coexistence of hippocampal sclerosis and another extrahippocampal epileptogenic entity, most commonly MCDs or acquired childhood lesions. 13 Satisfaction of search after detection of either HS or another epileptogenic entity can result in failure to detect both conditions, ultimately reducing success of epilepsy surgery and highlighting the importance of a systematic approach. 13
Multimodality imaging
Several of the entities discussed above have subtle findings that make detection difficult, even resulting in labeling as “normal.” These conditions include focal cortical dysplasias, small gangliogliomas, and DNETs. 13 Detection of these entities can be improved by selection of proper MRI techniques, incorporation of EEG and clinical findings, multimodality imaging with PET and SPECT, and a systemic approach for evaluating the epileptogenic lesions. Chart review should include age of seizure onset, seizure semiology, family history, and suspected localization from EEG findings. Multimodality imaging can also provide additional utility for epilepsy evaluation through confirmation or further mapping of the epileptogenic zone. In addition to being essential in the emergent setting for evaluation of acute hemorrhage, CT can also help detect conditions associated with calcifications. PET is helpful for detection of subtle FCD and surgical planning. SPECT can be used for interictal subtraction for localization, and fMRI is also commonly used for surgical planning. It is essential to avoid overinterpretation of structural lesions as epileptogenic foci without clinical correlation.
Systematic approach
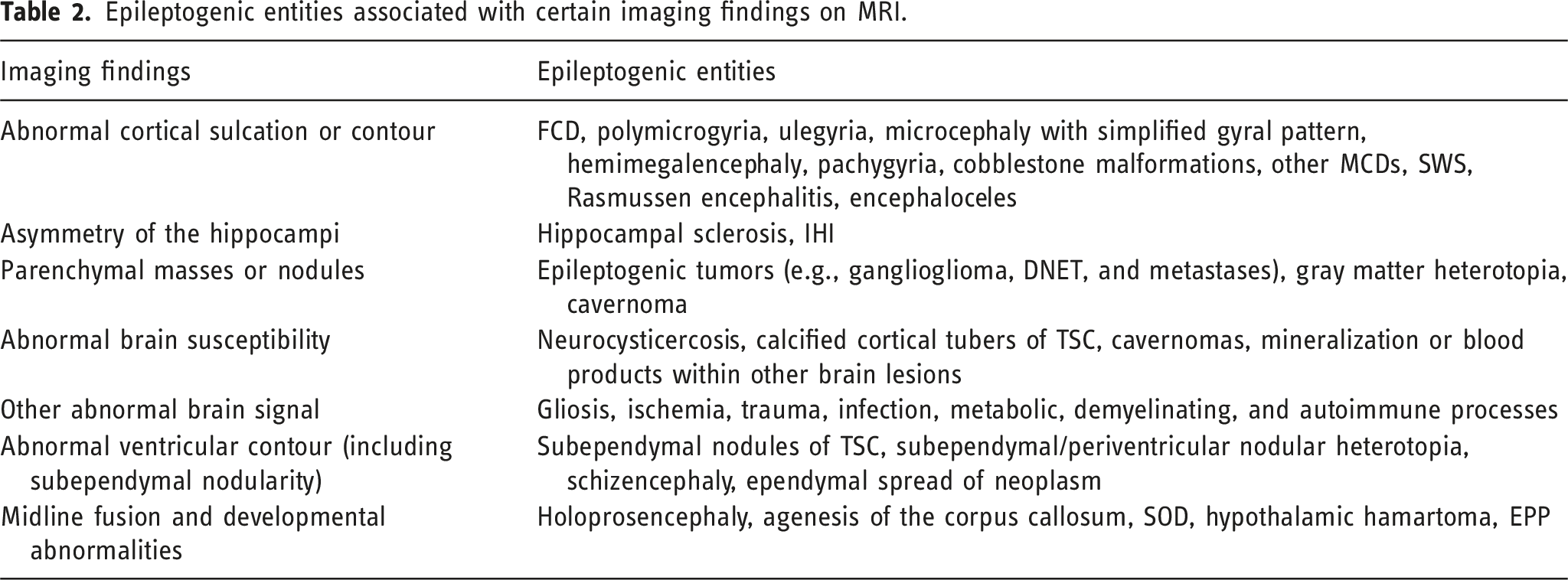
Epileptogenic entities associated with certain imaging findings on MRI.
Example reporting templates for pertinent positives and negatives in seizure imaging with differing styles.

Conclusion
Identification of epileptogenic foci can be facilitated with use of a systematic approach. Consideration of clinical semiology and electrophysiological information, recognition of the features of notable intracranial pathologies, use of specialized epilepsy protocols for certain relatively common conditions such as FCD and hippocampal sclerosis, and use of comprehensive epilepsy reporting templates can facilitate evaluation of structural causes of epilepsy on imaging.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.