
Abstract
Objective
Cryopreservation via vitrification of articular cartilage (AC) will increase the availability of graft tissue for treating large joint defects. To advance this research area, we compared the effects of 2 cryopreservation protocols in which 10-mm diameter human osteochondral dowels were cooled and stored in liquid nitrogen vapor.
Design
Dowels collected from healthy human knee joints (n = 3 donors) of deceased donors were randomly assigned to Protocol 8 (430 min) or Protocol 2BWF (410 min). Post-warming chondrocyte viability was assessed and normalized to fresh controls.
Results
Both protocols resulted in high chondrocyte viability after loading, vitrification, and rewarming (~80% of fresh control). Protocol 2BWF was subsequently used to vitrify and rewarm 3 human intact lateral femoral condyles. After rewarming, metabolic activity, normalized chondrocyte viability, histology, and matrix productivity were experimentally measured. Results documented ~82% of fresh chondrocyte viability post vitrification and rewarming, with similar results to the fresh control group on the other AC quality criteria.
Conclusion
These results demonstrate that both Protocol 8 and Protocol 2BWF can preserve the quality of vitrified human AC in osteochondral dowels and human intact femoral condyles.
Introduction
Articular cartilage (AC) defects are difficult to self-repair due to lack of blood vessels, nerves, and lymphatic supply, 1 and without proper treatment, large cartilage defects over 1 cm2 often progress into osteoarthritis (OA).2,3 Surgical procedures that are used to treat cartilage damage include osteochondral allograft transplantation, osteochondral autograft transfer, autologous chondrocyte implantation, and marrow stimulation techniques (subchondral drilling and microfracture).4 -6 Osteochondral allograft transplantation (a restorative cartilage method for symptomatic focal cartilage defects) has increasingly become the preferred procedure after cartilage repair surgery failures in large defects due to a well-developed, single-stage restorative cartilage procedure.7,8 However, this method is limited by the availability of fresh donor joints.9,10 Fresh AC is typically kept at 4 °C in a tissue bank for up to 28 days. Unfortunately, after 7–14 days of storage at this temperature, chondrocyte viability and matrix integrity begin to deteriorate.10 -12 These short holding times can lead to significant loss of priceless human tissue due to the inability to size and contour match and the logistics of having the patient and operating team available in addition to the time required for infectious disease testing. Long-term preservation of cells and tissues beyond the times achievable at hypothermic temperatures is possible with vitrification. Vitrification is an ice-free storage method developed for long-term storage of cells and tissues that can be achieved by superfast cooling or in the presence of high concentrations of cryoprotective agents (CPAs). 13
Previously, Jomha et al. 14 reported a successful vitrification protocol for intact human AC, reporting promising cell recovery with the use of a 4-step multi-CPA loading method that took 570 minutes. The long CPA loading protocol (570 min) was not ideal for practical use in tissue banks; therefore, a 420-minute (7-h) optimized protocol using mathematical modeling was developed.15,16 To extend this research, it was shown that the 7-hour CPA permeation strategy applied to the vitrification of intact porcine femoral condyle resulted in ~65–70% chondrocyte viability normalized to fresh control. 17 To extend these encouraging results in the direction of developing a reliable protocol that will work both in the lab and in clinical tissue banks, further research showed that adding a 10-minute half-strength CPA pre-exposure, adding sucrose (0.5 M) to the vitrification media in the last 2 steps of the optimized protocol, and performing CPA removal using descending concentrations of sucrose in the warming protocol (Protocol 8, 430 minutes) enhanced post-warming chondrocyte viability in AC. 18 However, a comprehensive study to examine the effects of these advances on human intact AC has not been performed. Moreover, translation of the technology from the laboratory to tissue bank has been always challenging due to differences in available material, equipment, and storage methods.
To address these challenges and lack of sufficient information on vitrification of human AC using our methods previously established in animals,16,19,20 the first experiment of the present study compared our last successful protocol (Protocol 8, 430 minutes) with a new protocol (Protocol 2BWF, 410 minutes) that was determined with mathematical modeling. The purpose was to evaluate their effectiveness in the vitrification of 10-mm diameter human osteochondral dowels while incorporating protocol advances from previous studies. In the second experiment, we cryopreserved intact human femoral condyles using the knowledge from the first experiment. Furthermore, we modified our vitrification-rewarming protocols by using cryobags as containers and storage in LN2 vapor to conform to tissue bank standard protocols. Achieving a successful vitrification protocol for different size of osteochondreal tissues is a good starting point for further optimization for future use in clinical applications involving vitrified human osteochondreal tissues, and could greatly increase the availability of donor tissue for transplantation.
Methods
Human articular Cartilage Preparation
Human research ethics approval (Pro00001141) was obtained from the University of Alberta Research Ethics Office. In the first experiment (dowel vitrification), healthy human knee joints (n = 3 donors) from deceased donors aged 56 (female), 40 (male), and 60 (male) were obtained from the Give Life Alberta Tissue Bank North (GLATBN) in Edmonton, AB, Canada, with consent from patients’ families to use donated cartilage for research. In the second experiment (intact condyle vitrification), healthy human knee joints (n = 3 donors) aged 43 (female), 47 (male), and 44 (male) were obtained from GLATBN. After tissue harvesting, knee joints were stored in 500 ml DMEM-F12 complete medium (Dulbecco’s Modified Eagle Medium Nutrient Mixture F12 [DMEM-F12; Gibco, NY, USA]) and transported to the research laboratory within 12 h of donor death. In the first experiment, 3 osteochondral dowels (2 dowels for vitrification and 1 dowel as fresh control) were cut from each sample with a sharp cutting device with a 10-mm diameter opening resulting in 10-mm diameter osteochondral dowels with full-thickness cartilage on a bone base to mimic the clinical scenario required for AC transplantation. The dowels were ~15 mm in height, of which 1–3 mm was AC and the rest was bone. In the second experiment, 3 lateral femoral condyles from the right knees were used and immersed in sterile DMEM-F12 supplemented with antibiotics [100 units/ml penicillin, 100 μg/ml streptomycin (Gibco)], and kept in the fridge at 4 °C for 6–12 hours prior to vitrification. Fresh control samples were taken from the same condyle and stored in sterile DMEM-F12 supplemented with antibiotics in the fridge at 4 °C until performing assessment techniques including membrane integrity stains (no longer than 4 hours after receiving tissue from CTC), metabolic activity, and pellet culture (up to 1 week). A minimum 80% absolute chondrocyte viability in the fresh controls before cryoprotectant exposure was used to screen out unhealthy cartilage donors.
Cryopreservation Protocols
Protocol 8
As described previously, Protocol 8 (430 min; Table 1 ), developed from the optimized protocol (420 min) 15 by adding a 10-minute Step 0 and adding sucrose (0.5 M) to the vitrification media in the last 2 steps of the optimized protocol and performing CPA removal using descending concentrations of sucrose in the warming protocol, enhanced post-warming chondrocyte viability in AC. 18
Protocol 8 and Protocol 2BWF That Incorporate Different Concentrations of CPAs at Different Vitrification Steps.

Protocol 2BWF
Protocol 2BWF ( Table 1 ) was developed for this work using mathematical modeling methods similar to those previously used in other studies.15,16 Namely, we used 1-dimensional Fick’s law to calculate cryoprotectant concentration profiles throughout a 2-mm-thick AC dowel during loading. We then used these profiles to compute solution freezing point and vitrifiability, using the multi-solute osmotic virial equation,21 -23 and a statistical model, 24 respectively. Protocol design was guided by the goal of achieving vitrifiability at the end of the last step while minimizing exposure time to the cryoprotectants and allowing each subsequent step to be performed at a lower temperature to reduce toxicity. The cryoprotectants used and their concentrations were chosen based on results of cryoprotectant toxicity studies,25,26 and existing protocols.
Figure 1 . shows the mathematical modeling results for Protocol 2BWF. As seen in panel (h), around half of the cartilage has exceeded the minimum score needed for vitrification by the end of the last loading step, while the other half has a score slightly below this cut off. Given that Fick’s law underestimates cryoprotectant concentration in comparison to the biomechanical triphasic model,27,28 it is likely that the actual vitrifiability of the cartilage is higher than the estimate calculated here.
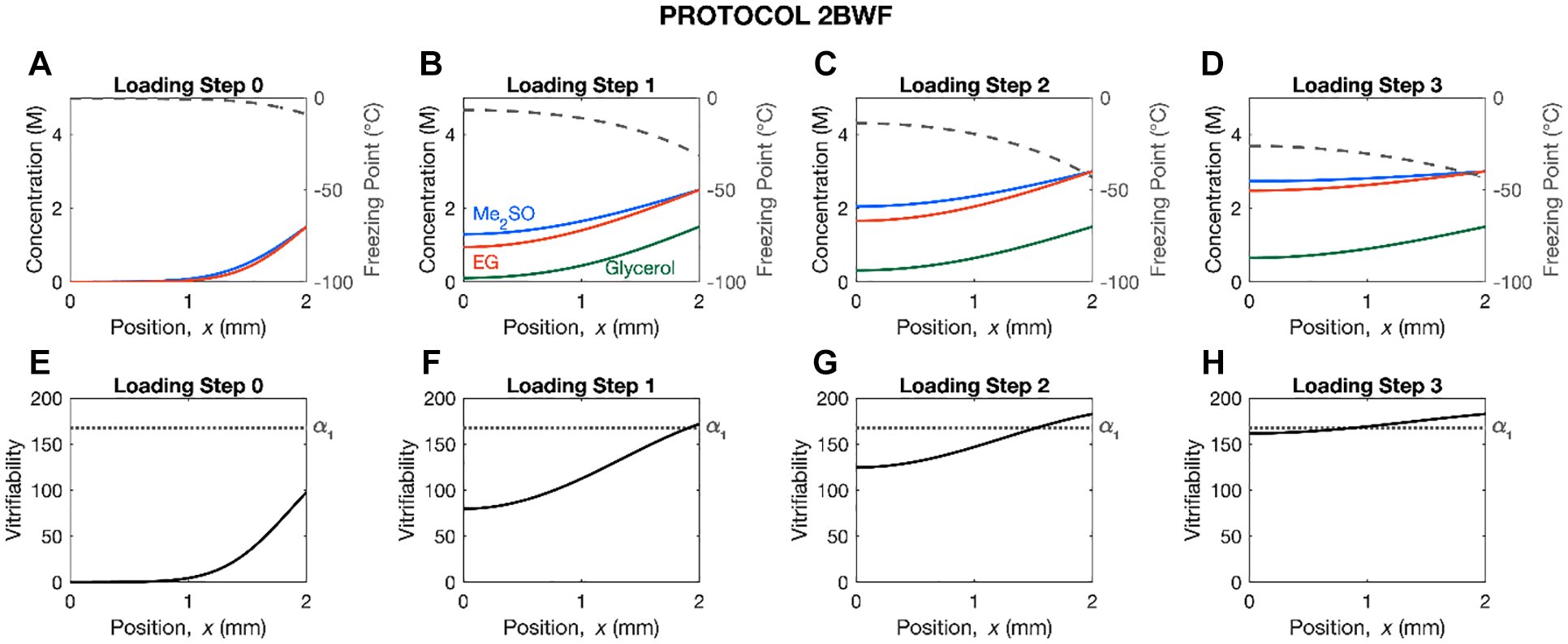
Mathematical modeling results of the cryoprotectant concentration, solution freezing point, and vitrifiability throughout a 2-mm-thick articular cartilage dowel at the end of each loading step of Protocol 2BWF. In panels (
Experiment 1. Evaluation of Effects of 2 Distinct Cryoprotective Agent Loading Protocols on Post-Warming Chondrocyte Viability in Vitrified 10-mm Human Osteochondral Dowels
Osteochondral dowels were randomly assigned to Protocol 8 and Protocol 2BWF as shown in Table 1 , which incorporate various CPA concentrations at various stages of the vitrification process. Vitrification media contained DMEM-F12 (Gibco, NY, USA) and CPAs, including dimethyl sulfoxide (Me2SO), ethylene glycol (EG), and propylene glycol (PG) (Thermo Fisher Scientific) plus chondroitin sulfate (CS; 0.1 mg/ml; Sigma-Aldrich Chemicals, St. Louis, MO, USA) and sucrose (0.5 M; Sigma-Aldrich Chemicals, St. Louis, MO, USA). DMEM-F12 was also used as the base media for the warming steps in both protocols. Sterile conical tubes (50 ml) were utilized as the containers for vitrifying osteochondral dowels. Each Step 0—Step 3 vitrification solution had a total volume of 50 ml, whereas the Step 4 vitrification solution had a total volume of 10 ml. Throughout the CPA loading process, the dowels were continuously stored in 50 ml conical tubes (Thermo Fisher Scientific; the tubes were placed in temperature-controlled methanol baths or an ice container), and after the CPA loading process was finished, the cartilage samples were put into cryobags (Bowers, Ontario, Canada; 1 dowel per bag) that contained 10 ml of Step 4 vitrification solution, which were swiftly sealed with a packaging machine after being vacuumed to remove the air. For rewarming we first placed the cryobag in a 37 °C water bath for 30 s, and once the glassy CPA surrounding the dowel melted, the dowel was quickly removed from the cryobag, patted dry and placed into 25 ml DMEM-F12 plus CS (0.1 mg/ml) and sucrose (1 M) for CPA removal at 4 °C for 30 min. The dowel was then placed in another 25 ml of fresh DMEM-F12 plus CS (0.1 mg/ml) and sucrose (0.5 M) for a second 30 minutes at 4 °C, followed by a third wash of 30 minutes at 4 °C in fresh DMEM-F12 plus CS (0.1 mg/ml). After warming, chondrocyte normalized viability was evaluated.
Experiment 2. Vitrification of Intact Human Femoral Condyle
In the first experiment, both Protocol 8 and Protocol 2BWF resulted in similar high chondrocyte viability after vitrification-warming (79.50 ± 2.53 and 80.78 ± 2.97% normalized to fresh control, respectively; P > 0.05). We chose Protocol 2BWF for vitrification of human intact femoral condyles (n = 3) because numerically it resulted in a slightly higher normalized chondrocyte viability in comparison with Protocol 8 (though not statistically significantly different) and it is 20 minutes shorter. Except for the type of the container, the amount of media and a few details of the rewarming, we followed the same procedures for vitrification and warming of intact femoral condyle as we did in the first experiment. Sterile cups (250 ml; Corning, NY, USA) were utilized as the containers for vitrifying osteochondral dowels due to the size of the tissue. Each Step 0 − Step 3 vitrification solution had a total volume of 200 ml, whereas there was no Step 4 vitrification solution in this experiment. A vacuumed cryobag was used (no surrounding Step 4 vitrification solution) after CPA loading steps based on previous experiments (Wu et al. 17 see Discussion). Cryobags were kept in liquid nitrogen vapor for 1 week. For rewarming we first placed the cryobag on the bench (22 °C) for 10 minutes. Then the cryobag was placed in a 37 °C water bath for 90s, and once the glassy CPA surrounding the condyle melted, the condyle was quickly removed from the cryobag, patted dry and placed into 200 ml DMEM-F12 plus CS (0.1 mg/ml) and sucrose (1 M) for CPA removal at 4 °C for 30 min. The condyle was then placed in another 200 ml of fresh DMEM-F12 plus CS (0.1 mg/ml) and sucrose (0.5 M) for a second 30 minutes at 4 °C, followed by a third wash of 30 minutes at 4 °C in fresh DMEM-F12 plus CS (0.1 mg/ml). After warming, chondrocyte normalized viability, metabolic activity, and matrix production by histology/immunohistochemistry were evaluated.
Assessment of Articular Cartilage chondrocyte viability via cell membrane integrity stain
A vibratome (Leica VT1000 S) was used for sectioning cartilage slices of 85-μm thickness from each osteochondral dowel cutting from the articular surface to the bone-cartilage junction and cartilage slices were placed in PBS until staining. To label membrane-damaged (dead cell, red color) and membrane-intact (live cell, green color) chondrocytes, a mixture of two fluorescent dyes: 9 μM propidium iodide (Sigma) and 6.25 μM Syto 13 (Molecular Probes) were used. Slices of cartilage were put on microscope slides, stained with a dye solution, then covered with a coverslip, and incubated in the dark for 15 minutes. Fluorescein isothiocyanate and tetramethylrhodamine isothiocyanate (FITC-TRITC) dual filters were used with the following spectra peak maxima wavelengths: 488 nm/503 nm and 535 nm/617 nm (excitation/emission) to capture images using a Nikon TiS inverted microscope. The image processing software used was Adobe Lightroom 4.3. Viability 3.2 (Locksley McGann, University of Alberta) was used to calculate the viability of the cells. The chondrocyte viability after vitrification was normalized to its own fresh control before CPA loading. The formula below was used to calculate the normalized cell viability of the experimental samples:
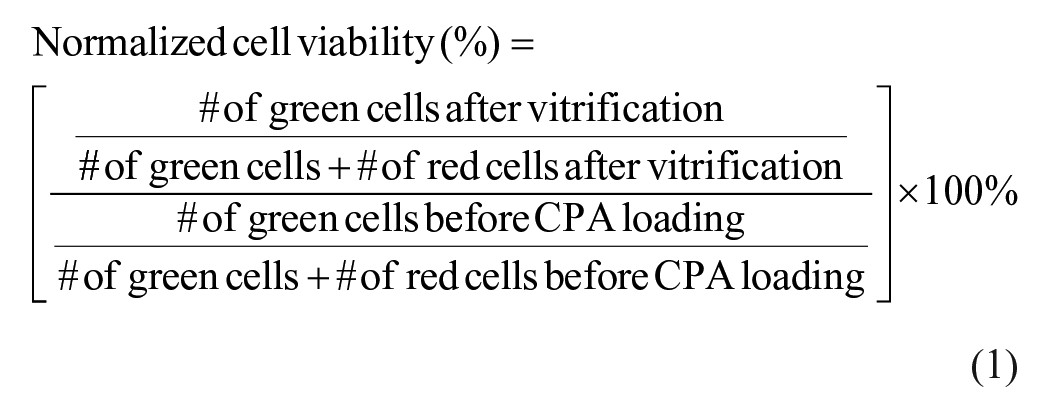
Chondrocyte metabolic activity
AlamarBlue was used to measure the metabolic activity of chondrocytes, as described before. 14 As a fluorescence indicator, AlamarBlue relies on the reduction reaction of metabolically active cells to transform the nonfluorescent blue resazurin into the highly fluorescent red resorufin. After tissue warming and the removal of CPA, cartilage from intact femoral condyles was removed, weighed, and washed in sterile PBS with antibiotics. AC that had not been vitrified or subjected to CPAs served as the positive controls (fresh), whereas cartilage that had been submerged in liquid nitrogen without CPAs served as the negative controls. AC samples were incubated with the alamarBlue assay solution [5 ml DMEM-F12 supplemented with 10 ng/ml transforming growth factor beta 1, 100 nM dexamesasone, 0.1 mM ascorbic acid, and 500 μl alamarBlue (Invitrogen, Burlington, Canada)]. Samples were incubated at 37 °C for 8 days, with fluorescence readings taken every 24 hours using the Cytofluor 2.0 program. The wavelengths of the fluorescence readings were tuned to 485 nm for the excitation and 580 nm for the emission, with a gain of 45. Every sample in the experimental group had its fluorescence assessed after 24 hours, 48 hours, 72 hours, 96 hours, 120 hours, 144 hours, 168 hours, and 192 hours. Readings of blank (standard) samples (alamarBlue solution with no cartilage) were subtracted from values of the experimental samples to generate a value in relative fluorescent units (RFU) divided by weight of the sample. The formula below was used to calculate the normalized RFU values of the experimental samples:

Assessment of matrix production using histology/immunohistochemistry
Chondrocyte isolation/recovery/collection
After warming, up to 0.5 g of cartilage were shaved off from each condyle, and then digested by 0.15% collagenase solution (filtered, Worthington) for 22 h. Once the cartilage digestion was finished, the chondrocytes were collected by centrifugation, and then resuspended in DMEM-F12 for chondrocyte recovery for 72 h at 37 °C. After chondrocyte recovery, trypsin-EDTA solution (Gibco) was used to disassociate chondrocytes, and then chondrocytes were collected for cell counting via centrifugation. Once the chondrocytes were resuspended in DMEM-F12 complete medium, pipetting was used to combine 15 μl of the cell suspension with 15 μl of trypan blue (Sigma-Aldrich Chemicals, St. Louis, MO, USA). For the purpose of chondrocyte counting, 10 ml of this mixture were carefully put in a hematocytometer using a pipette. The cell count was calculated by adding the counted cells in 4 areas of equal size, dividing by 4, and then multiplying by the total volume of chondrocyte suspension solution and a dilution factor of 10,000. After chondrocyte counting by trypan blue, 5 × 105 chondrocytes were resuspended in defined chondrogenic SFM [DMEM-F12, HEPES (10 mM), human serum albumin (125 mg/ml), dexamethasone (100 nM), ascorbic acid 2-phosphate (365 lg/ml), and L-proline (40 lg/ml) (Sigma-Aldrich), ITS +1 premix (5 μl, 100×) (Corning, Discovery Labware, Inc.), TGF-b3 10 ng/ml; ProSpec, NJ, USA, 100 units/ml penicillin, 100 μg/ml streptomycin; Cytiva HyClone, Ca, Montreal], and after being centrifuged at 433 × g and 22 °C for 5 min, the resulted pellets were cultured for 21 days under 5% CO2 and 3% O2 at 37 °C in a humidified incubator, with the culture media changed twice a week.
Pellet processing
After a 21-day culture, pellets were rinsed with sterile PBS (Ca2+/Mg2+ free) and were fixed with 10% formalin for 24 h before paraffin embedding. A microtome (Leica) was used to prepare pellet sections with thicknesses of 5 μm, followed by section drying at 37 °C overnight in a dry incubator.
Safranin O and fast green staining
After deparaffinization and rehydration, sections were stained with 0.02% fast green (Sigma-Aldrich Chemicals, St. Louis, MO, USA) and 0.1% Safranin O (Sigma-Aldrich Chemicals, St. Louis, MO, USA) for 4 minutes and 15 minutes, respectively before mounting on slides for imaging.
Collagen type I/II immunolocalization
The same processes for fixation, dehydration, embedding, sectioning, and mounting on slides were performed to localize collagen type I/II. The slides were treated with 1 mg/ml of protease XXV (Thermo Fisher Scientific) at 22 °C for 30 minutes and then with 1 mg/ml of hyaluronidase (Sigma) at 37 °C for 30 minutes. Primary antibodies for collagen type I (CL50111AP-1; Cedarlane) and collagen type II (IIII6B3; DSHB) were applied on slides and after being blocked with 5% bovine serum albumin (Cell Signaling Technology), slides were incubated overnight at 4 °C. After washing slides in PBS, secondary antibodies (goat anti-rabbit IgG Alexa Fluor 594; 150117; Abcam and goat anti-mouse IgG Alexa Fluor 488; 150080; Abcam) for collagen types I and II, respectively were added to slides and incubated for 30 minutes at 4 °C. DAPI (2.9 mM; Sigma-Aldrich Chemicals, St. Louis, MO, USA) was added to slides (for 5 minutes) after washing the slides in PBS, and then mounted with glycerol before fluorescent microscopy. A Nikon ECLIPSE Ti-5 inverted fluorescence microscope and a Nikon DS-Fi2 digital camera were used to image the sections. The cartilage sections (live-dead; collagen types I and II) were imaged using dual filters for FITC-TRITC with the following spectral peak maximum wavelengths: 488 nm/503 nm and 535 nm/617 nm (excitation/emission).
GAG/DNA measurement
A dimethylmethylene blue (DMMB) assay was used to determine the GAG content of pellets. Before usage, pellets were weighed, rinsed with PBS, and placed in a freezer at −80 °C. Utilizing a dry block heater (Thermo Fisher Scientific), pellets were heated and then digested in 250 μl of 1 mg/ml proteinase K for an overnight period at 56 °C. The controls for a standard curve were PBE/cysteine buffer (10 mM Na2EDTA, 100 mM Na2HPO4, pH = 6.5, 1.75 mg/ml cysteine, Sigma) and 0–100 g/ml chondroitin sulfate A sodium salt (CS). With an increasing concentration of CS, the standard curve was created in eight 100 μl Eppendorf tubes. After protein digestion, a 5 μl aliquot of digested cartilage was pipetted in triplicate into an ultraclear 96-well plate (NUNC, Thermo Fisher Scientific). Each digested cartilage sample was combined with 5 μl of PBE/cysteine buffer before being diluted 1:50 with 250 μl of DMMB dye (Sigma-Aldrich). Data were standardized to the blank reading of H2O (260 L, without DMMB) and the CS standard controls for each plate after being read at 525 nm. The CyQUANTTM proliferation assay kit for cells in culture (Invitrogen, ON, Canada) was used to measure DNA content. A 5-μl cartilage sample was pipetted into a 96-well plate in triplicate after digestion with 1 mg/ml proteinase K. To make the total volume equal to 200 μl in each well, 195 μl of working buffer was added. Using an assay kit, 29 DNA solutions and working buffer were created. 50 μl of CyQUANT GR dye, 1 ml of cell-lysis buffer, and 19 ml of Milli-Q water were combined to make 20 ml of working buffer. The plates were read using the spectra peak maxima for 530/25 nm emission and 450/50 nm excitation, with the provided bacteriophage λDNA serving as a standard reference.
Statistical Analysis
Means ± standard deviations (SD) were used to report numerical data. Using IBM SPSS 28.0 software, a 2-way analysis of variance (ANOVA) with Tukey’s multiple comparison tests was performed to identify any variations in normalized cell viability between experimental groups at each trial. Statistical significance is noted when P < 0.05. All figures were plotted in Microsoft Excel (V. 16.61.1).
Results
Evaluation of Effects of 2 Distinct Cryoprotective Agent Loading Protocols on Post-Warming Normalized Chondrocyte Viability in Vitrified 10-mm Osteochondral Dowels
In this experiment, healthy human knee joints (n = 3 donors) from deceased donors aged 56, 40, and 60 years (mean ± SD: 52.3 ± 10.58 years) were used. Figure 2A and B displays representative fluorescence images of cartilage slices from osteochondral dowels from (A) the experimental group using Protocol 2BWF, and (B) the experimental group using Protocol 8 following vitrification-rewarming. As shown in Figure 2C , both Protocol 2BWF and Protocol 8 resulted in a relatively high normalized chondrocyte viability (79.50 ± 2.53 and 80.78 ± 2.97%, respectively), following vitrification-rewarming. However, there was no statistically significant difference between the effects of 2 CPAs loading-vitrification protocols on the normalized chondrocyte viability (P > 0.05).
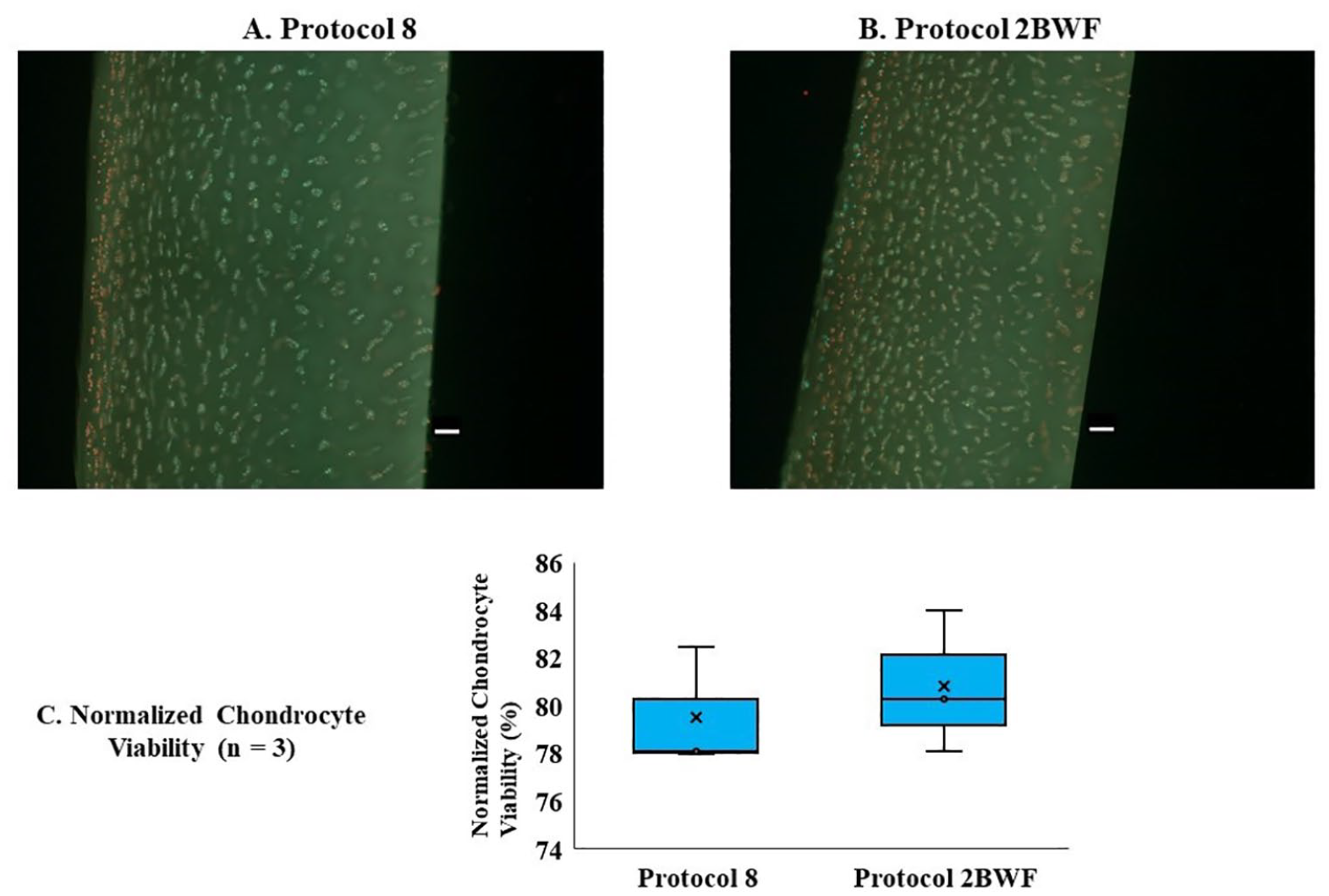
Chondrocyte viability of 10-mm osteochondral dowels after vitrification using 2 CPA loading-vitrification protocols. Representative images of chondrocyte viability in AC slices (
Post-Warming Normalized Chondrocyte Viability and Metabolic Activity of Intact Femoral Condyle
In this experiment, human knee joints (n = 3 donors) from deceased donors aged 44, 47, and 43 years (mean ± SD: 44.66 ± 2.08 years) were used. Figure 3A displays representative fluorescence image of cartilage slices from whole femoral condyles after vitrification-rewarming with Protocol 2BWF. As shown in Figure 3B , normalized chondrocyte viability of the vitrified-rewarmed intact femoral condyles using Protocol 2BWF was 82.90 ± 10.58%. Figure 4A displays representative photos of the chondrocyte metabolic activity of both fresh cartilage and cartilage after vitrification-rewarming of the entire femoral condyle with Protocol 2BWF from 24 to 192 hours after rewarming. After 48 hours of cartilage rewarming, the fresh control group showed a higher (P < 0.01) normalized chondrocyte metabolic activity than that of the vitrified-rewarmed group and the negative control group. However, vitrified-rewarmed chondrocyte metabolic function began to normalize 72 hours after warming and was similar (P = 0.054) to the fresh control group, with very similar metabolic activity noted by 96 hours after warming (P = 0.11) which was maintained until 192 hours (P = 0.57) after rewarming (see Fig. 4B ).
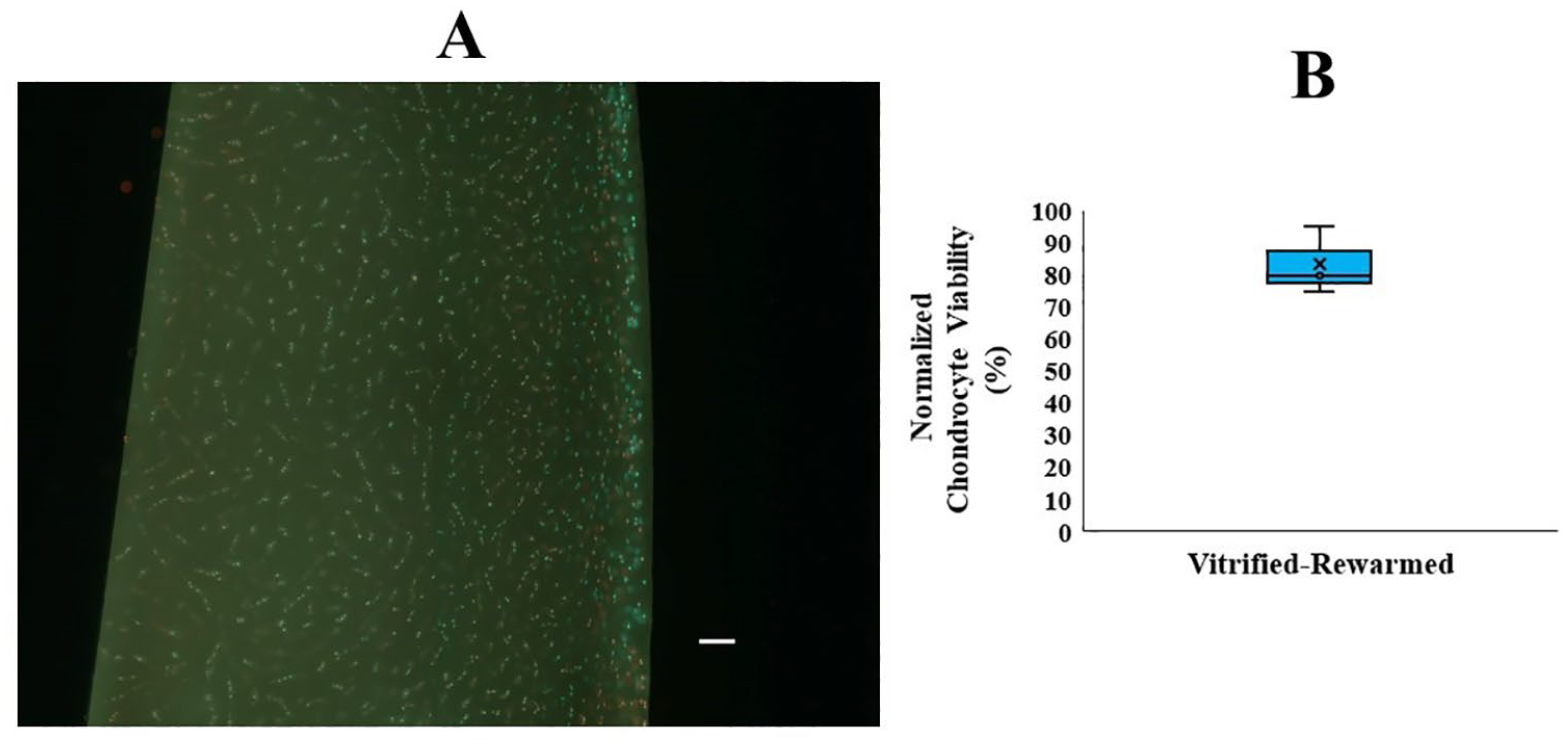
Intact femoral condyle chondrocyte viability after vitrification-rewarming.
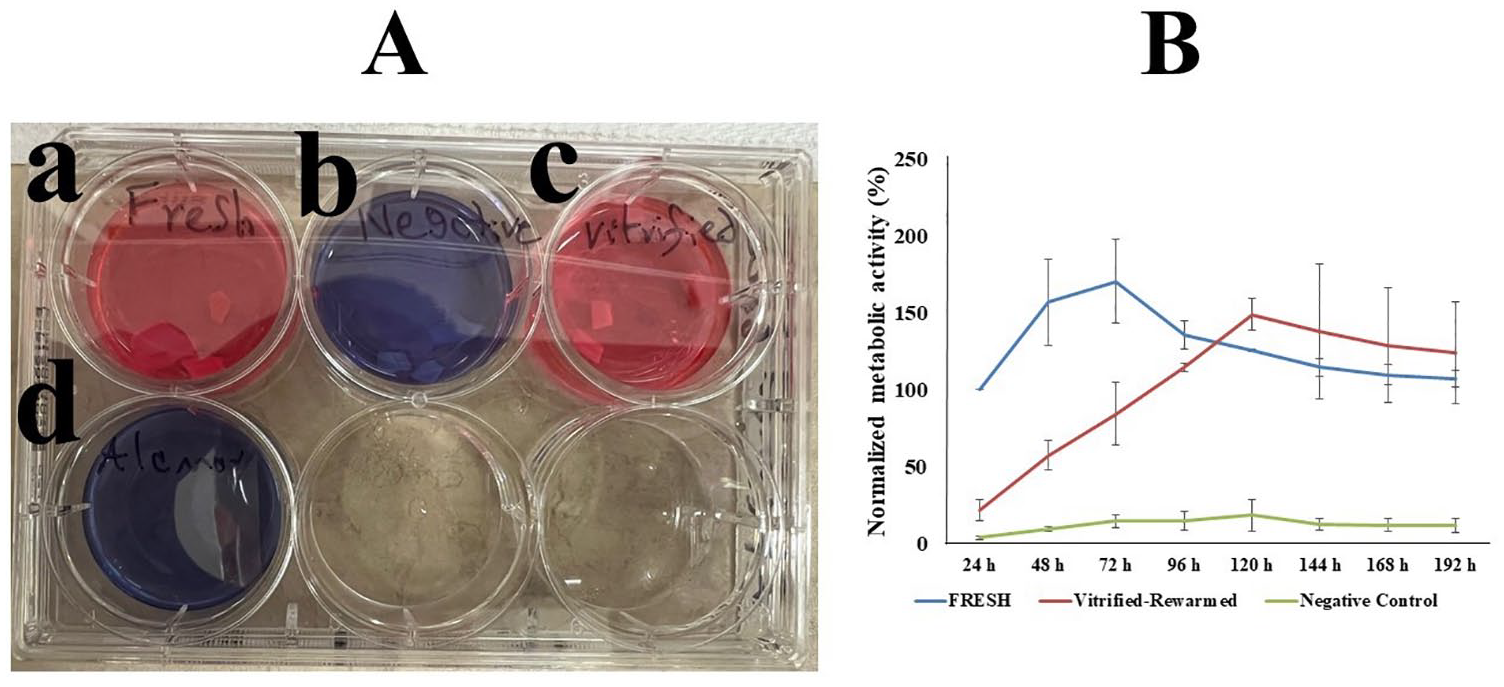
Intact femoral condyle chondrocyte metabolic activity after vitrification-rewarming with Protocol 2BWF. (
Assessment of Matrix Production After Vitrification-Rewarming of Intact Femoral Condyle
Safranin O staining was used to determine the presence of proteoglycan [sulfated glycosaminoglycans (GAGs)] of cartilage sections after 3 weeks of culture ( Fig. 5 ). In order to keep healthy cartilage intact, GAG production is crucial. For both the vitrified group and the fresh control group, the same pink color (Safranin O) pattern was seen ( Fig. 5A ). In order to evaluate the expression of collagen in both fresh control and vitrified-rewarmed cartilage pellets following a 3-week culture period, cartilage slices were immunostained for collagen types I and II ( Fig. 5B ). Both fresh control and vitrified-rewarmed pellets were immunopositive for collagen type I and II and showed similar expression patterns. To evaluate the cell density of fresh control and vitrified-rewarmed cartilage, DAPI staining ( Fig. 5C ) was used. The results showed a similar cell density and functionality for the fresh control and vitrified-rewarmed pellets after the 3-week culture period. Chondrocyte synthesis functionality which was assessed by the amount of GAG per DNA showed that after the 21-day culture period, the vitrified-rewarmed full femoral condyle had higher (p = 0.015) GAG/DNA content than the fresh control group ( Fig. 5D ).
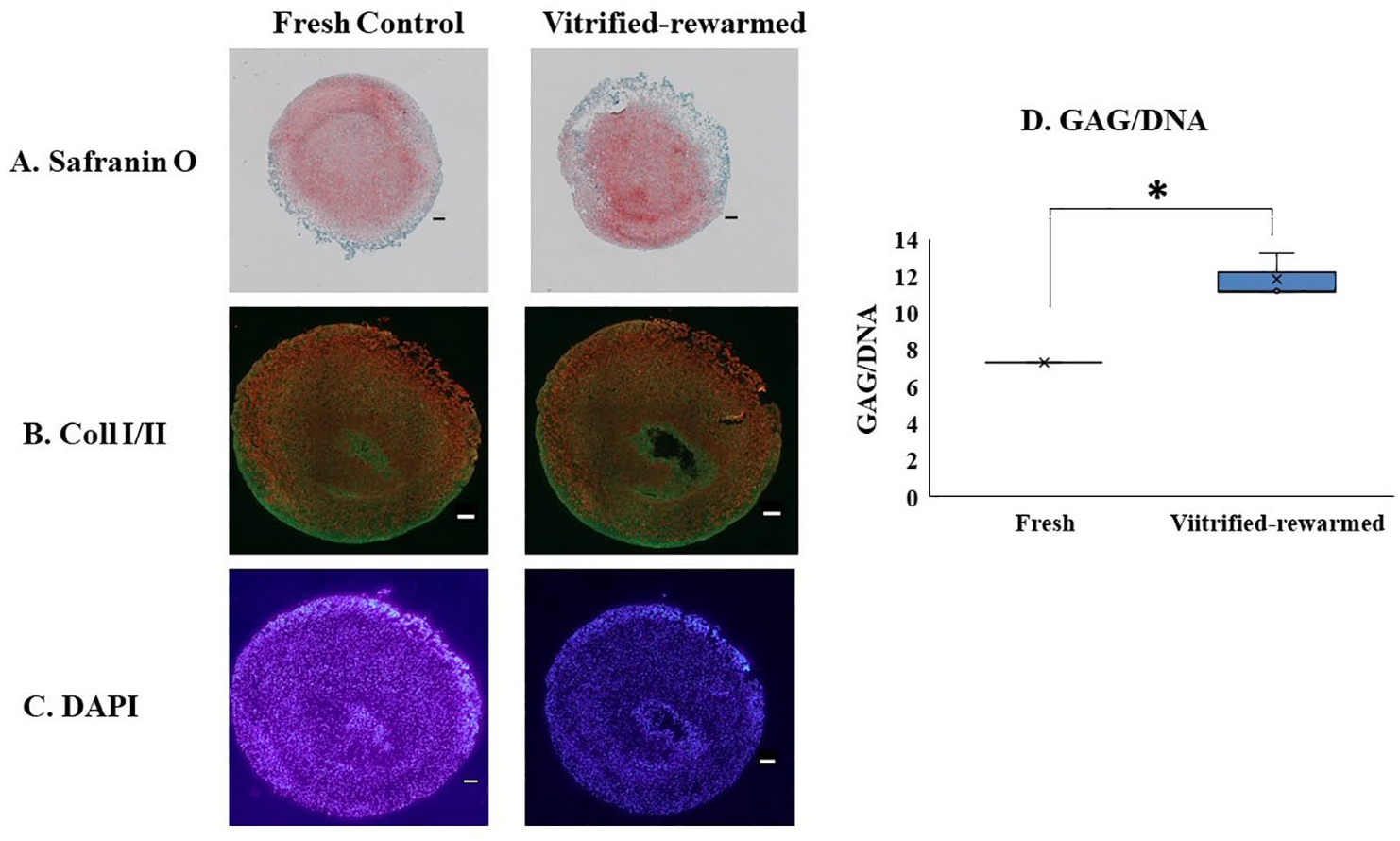
Assessment of matrix productivity and immunofluorescent staining of cartilage pellets from human intact femoral condyle post vitrification-rewarming. (
Discussion
In this study, we effectively vitrified and stored human 10-mm osteochondral allografts at cryogenic temperature by using 2 new CPA loading-vitrification-rewarming protocols. Compared to the existing standard of hypothermic storage at 4 °C for 28 days, our strategy extends the time available for clinical use indefinitely given that biologic reactions are negligible at the temperature of liquid nitrogen vapor, giving surgeons more time to schedule procedures. In the first experiment of our study, 2 progressive vitrification-rewarming techniques (Protocol 8 and Protocol 2BWF) were validated using 10-mm human osteochondral dowels, delivering a positive result that satisfies the 70% chondrocyte viability criteria for clinical transplantation.7,10
Protocol 8 and Protocol 2BWF resulted in ~80% of fresh chondrocyte viability post vitrification-rewarming. This is consistent with a previous study 30 that demonstrated that the chondrocyte viability of human particulated AC declined and resulted in ~80% of fresh chondrocyte viability after vitrification which was a greater decline than that of porcine samples where normalized viability was ~90% of fresh chondrocyte viability. The quality of the donated human sample may be the cause of this increased viability decline in the vitrified-rewarmed groups. Donor age is a variable that could have a significant impact on the cartilage quality and ability to withstand the CPA exposure and vitrification process. In the present study, human AC was obtained from 3 donors (ages 56, 40, and 60), ages at which cartilage quality deterioration had likely begun even if at a microscopic level. This is clearly over the expected age of human donors for AC transplantation in the clinical setting. When compared to AC in children under the age of 13 30 proteoglycans begin to degrade and chondrocyte density declines as age grows to 50–60 years. 31 With increasing frequency and intensity of everyday exercise over several decades, elderly chondrocytes gradually undergo slow apoptosis that results in a large loss of water content in the cartilage matrix. 32 Another important variable leading to reduced chondrocyte viability of human vitrified-rewarmed tissues in the current study as compared to pig tissues in previous studies is the processing time of human AC after donor death. An earlier study has shown that longer storage after harvest (24 h) at 4 °C compared to shorter storage (less than 6 h) resulted in increased sensitivity to the harmful effects of high CPA concentrations. 33 This could be the result of the reactive oxidative species created during the process of loading CPAs, 34 and a possible enhanced detrimental effect on stressed chondrocytes. In the current experiment, the time from donor death to harvest to vitrification protocol was approximately 12 hours. There were no significant differences in normalized chondrocyte viability when comparing Protocols 8 and 2BWF for vitrification of 10-mm human osteochondral dowels. The high normalized cell viability using Protocol 8 for the human tissue in this study was consistent with a previous study using procine AC. 18 That study demonstrated that supplementing sucrose in the vitrification media for the last 2 steps and removing CPA using decreasing amounts of sucrose in the warming media (Protocol 8) enhanced post-warming normalized chondrocyte viability in 6.9-mm diameter porcine osteochondral dowels vitrified with our previously developed protocol. Protocol 2BWF also demonstrated high normalized cell viability in human tissue similar to Protocol 8.
Protocol 2BWF was used in the second experiment of this study to vitrify human intact femoral condyles because the normalized chondrocyte viability in Protocol 2BWF was slightly higher (though not statistically significantly different) than in Protocol 8 (80.78% vs. 79.50%, respectively), and 20 minutes shorter (410 min vs. 430 min, respectively), although the other Protocol likely would have performed similarly. In the second experiment of this study, a vacuumed cryobag was used (no surrounding Step 4 vitrification solution) after CPA loading steps based on previous experiments. Wu et al. 17 previously showed that having a solution around the cartilage tissue can be associated with thermal cracking, and it slows down the cooling and warming of the cartilage, which can cause tissue devitrification and chondrocyte mortality. In the second experiment of this study, Protocol 2BWF resulted in ~82% of fresh chondrocyte viability post vitrification-rewarming, and no discernible or statistically significant differences were observed between human intact femoral condyles vitrified with Protocol 2BWF and the fresh control group when comparing metabolic activity, cell density, and proteoglycan and collagen type I & II expression within the extracellular matrix with the exception of a higher GAG/DNA ratio in the vitrified group.
Taken together, these results demonstrate that Protocol 2BWF is a successful vitrification protocol for human intact AC located on femoral condyles. The used vitrification protocol not only demenstrated normalized viability above 80%, but also the non-normalized viabilities were above 70% which met the minimum recommended rate (70%) for transplantation. At such low subzero temperatures in LN2 vapor, biochemical reactions cease and cellular processes stop thereby preventing any aging or injury from occurring. Tissue can be stored in this condition indefinitely. Furthermore, metabolic activity is maintained after a 2–3 day warm up period. Finally, the cells that survived the vitrification process continue to act like normal articular chondrocytes producing sulfated GAGs and collagen type II (along with collagen type I as seen in the fresh controls which occurs when chondrocytes are removed from their matrix).
The vitrification protocol used in this experiment was designed to enable a move to clinical translation in tissue banks. Therefore, a number of variables were incorporated that were not present in our previous protocols using human tissue. One concern has been loss of chondrocytes in the superficial zone possibly due to significant osmotic effects with the rapid addition of high concentrations of CPAs in the first step. 18 To counter this, we used an initial step at a lower starting CPA concentration (Step 0). This was intended to prevent osmotic damage and appears to have been effective as few superficial cells were disrupted and possibly even less damage would have been noted if younger, healthy tissue could have been used in this experiment. To augment the vitrifiability of the solutions, CPA loading Steps 3 and 4 had sucrose supplementing the CPA loading-vitrification media. Sucrose was also added into the warming solution to minimize osmotic effects as the CPAs were diluted out of the cartilage. According to reports, sucrose helps penetrating CPAs promote the extracellular medium’s vitrification state, significantly lowering the production of extracellular ice that is damaging to cells. 35 Furthermore, excessive cell lysis and swelling could be prevented by including sucrose in the rewarming media as sucrose aids in regulating the amount of water that enters cells during CPA removal.35,36 In vitrified-rewarmed human intact femoral condyles, these synergistic actions appear to enhance post-warming chondrocyte quality.
Compared to the initial published protocol, 14 we have altered the combination of CPAs used as well as the length of exposure in an effort to decrease the time to vitrify resulting in decreased cell toxicity due to lessened CPA exposure. 15 Furtheremore, we modified the rewarming protocol by adding a rewarming step at room temperature before water bath warming, and using descending concentrations of sucrose in the washing media. These modifications enhanced post-warming normalized chondrocyte viability in human intact femoral condyle vitrified with our new CPA loading-vitrification and warming protocol (Protocol 2BWF) compared to the previously developed protocol 17 for vitrification of porcine intact femoral condyles (~80% vs. ~65%, respectively). These adjustments were used to reduce unequal thermal expansion causing thermo-mechanical stress in large tissue samples.17,37 Other adaptations to facilitate the translation of the technology from laboratory to tissue banks included the use of a cryobag instead of a fixed container. This allows for a more compact storage method and decreases the use of excess CPAs. Finally, storage in LN2 vapor was used as this is the standard storage method (as opposed to in LN2 itself) in our local tissue bank (GLATBN, Edmonton, Canada). It is worth mentioning that after vitrification-warming and CPA removal, all 3 intact condyles demonstrated visible fractures in the cartilage surface. This could be due to differential warming rates resulting in mechanical stresses. This cracking phenomenon requires further optimization to ensure that any tissue prepared for transplantation remains structurally intact.
In conclusion, results of the present study demonstrate the ability to scale up the vitrification of human AC from 10-mm osteochondral dowels to human intact femoral condyles using our previous knowledge related to different CPA loading protocols and rewarming strategies combined with new strategies to overcome reported challenges in vitrification of intact femoral condyles. We demonstrated that both our CPA loading-vitrification protocols (Protocol 8 and Protocol 2BWF) resulted a high (~80%) normalized chondrocyte viability post vitrification in 10-mm human osteochondral dowels. In human intact femoral condyles, we showed that the quantity and quality of chondrocytes using Protocol 2BWF was equivalent to that of the fresh control, an essential result for effective clinical implementation and use. In addition, we incorporated variables such as usage of cryobags and storage in LN2 vapor to facilitate translation to clinical tissue banks.
Footnotes
Acknowledgments and Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the Edmonton Orthopedic Research Committee. JAWE holds a Canada Research Chair in Thermodynamics. We extend our sincere gratitude to Dr. Locksley E. McGann for supplying the Viability 3.2 program. Human tissues were provided by the Give Life Alberta Tissue Bank North (GLATBN), Edmonton, Canada.
Author Contributions
MS, JAWE, and NMJ designed and advised on the experiments; MS performed lab bench experiments, data acquisition, and data analysis; SC performed mathematical modeling and designed the vitrification protocols; and MS wrote the manuscript with guidance from NMJ and JAWE. All authors reviewed and revised the manuscript for submission.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: NMJ and JAWE are co-inventors on the following patent: N. M. Jomha, L. E. McGann, J. A. W. Elliott, G. Law, F. Forbes, A. Torghabeh Abazari, B. Maghdoori and A. Weiss, “Cryopreservation of articular cartilage,” US 8,758,988 & CA 2,778,202. On behalf of all authors, the corresponding author states that there are no other conflicts of interest.
Ethical Approval
Human research ethics approval was obtained from the University of Alberta Research Ethics Office.
Availability of Data and Materials
The data used to support the findings of this study are available from the corresponding author upon request.