
Abstract
Objective
It is unknown why some athletes develop chondromalacia and others do not, even when accounting for similar workloads between individuals. Genetic differences between individuals may be a contributing factor. The purpose of this work was to screen the entire genome for genetic markers associated with chondromalacia.
Design
Genome-wide association (GWA) analyses were performed utilizing data from the Kaiser Permanente Research Board (KPRB) and the UK Biobank. Chondromalacia cases were identified based on electronic health records from KPRB and UK Biobank. GWA analyses from both cohorts were tested for chondromalacia using a logistic regression model adjusting for sex, height, weight, age of enrollment, and race/ethnicity using allele counts for single-nucleotide polymorphisms (SNPs). The data from the 2 GWA studies (KPRB and UK Biobank) were combined in a meta-analysis.
Results
There were a total of 3,872 combined cases of chondromalacia from the KPRB and the UK Biobank cohorts. Genome-wide significant associations with chondromalacia were found for rs144449054 in the ARHGAP15 gene (OR = 3.70 [2.32-5.90]; P = 1.4 × 10−8) and rs188900564 in the MAGEC2 (OR = 2.07 [1.61-2.65]; P = 3.7 × 10−9).
Conclusions
Genetic markers in ARHGAP15 and MAGEC2 appear to be associated with chondromalacia and are potential risk factors that deserve further validation regarding molecular mechanisms.
Introduction
Articular cartilage is a highly specialized tissue that provides a low-friction gliding surface within joints. 1 Unfortunately, damage to this surface can occur as people age, leading to chondromalacia of the joint and eventual symptomatic osteoarthritis, with the most commonly effected joint. 2 Chondromalacia, or damage to the articular cartilage of a joint, is common even among young individuals. For those below 40 years of age undergoing magnetic resonance imaging in uninjured knees, up to 17% were noted to have imaging evidence of chondromalacia. 3 Furthermore, full thickness cartilage lesions were noted in 36% of competitive athletes, with 40% of this group participating in professional sport. 4
One of the main causes of articular cartilage defects is joint trauma, with meniscus and ACL injury being well-known causes of chondromalacia within the knee. 5 Other factors such as age, joint malalignment, obesity, and overuse can contribute to the development of chondromalacia as well as osteoarthritis within a joint. 6 In athletes, however, many of these later factors are not relevant and chondromalacia develops outside of any known injury. Therefore, there may be other causative biological factors.
An attractive hypothesis is that genetic variation partly accounts for individual differences in the susceptibility to chondromalacia. Prior studies have utilized genome-wide association (GWA) screens to investigate for genetic risk factors associated with other musculoskeletal conditions, but there have been no studies of genetic differences associated with chondromalacia. The advantages of a GWA screen are that it reports the strongest signals from across the entire genome, and the criteria for statistical significance are well developed which aids in reproducibility in validation studies. The main disadvantage of GWA studies—that large cohorts are required to achieve statistical significance (P < 5 × 10−8) to account for the large number of tested polymorphisms (multiple hypothesis correction)—can be overcome with large sample size.
The purpose of this study was to perform a screen of the entire genome for polymorphisms associated with chondromalacia using data from 2 large cohorts containing hundreds of thousands of participants. We hypothesized that genetic differences would be present between those patients with a documented diagnosis code for chondromalacia versus those without.
Methods
Genome-wide association analyses (GWAS) for chondromalacia were performed using data from the KPRB (with which the Kaiser Permanente, Northern California Research Program in Genes, Environment and Health [RPGEH] is affiliated) and from the v3 release of UK Biobank. This study analyzed stored data from KPRB and UK Biobank participants who consented to genomic testing and use of their genomic data, as well as health data from the KPNC and UK Biobank electronic health records. The health and genotype data for the participants were de-identified. All study procedures were approved by the Institutional Review Board of the Kaiser Foundation Research Institute. This study analyzed stored data from UK Biobank subjects who consented to genomic testing and use of their genomic data. The health and genotype data for the subjects were de-identified.
KPRB Cohort
KPRB is an integrated healthcare delivery organization, which has an active membership of 3.5 million people. 7 Comparisons with the general population have shown that the membership is a representative of the population of Northern California, with the exception of extremes of the socioeconomic spectrum. In 1995, KPRB instituted a comprehensive electronic health system, which records physician diagnoses, prescriptions, and lab results from all Kaiser inpatient and outpatient encounters. KPRB has high membership retention, with over 90% of those above age 65, and 66% of all active members as of June 2012, having 5 or more years of retrospective membership.
Our analysis cohort includes 83,414 individuals of European ancestry who were genotyped at 670,572 SNPs using Affymetrix Axiom genome-wide arrays. Genotypes were pre-phased with Shape-IT v2.r644 (accessed February 2, 2016) then imputed to a cosmopolitan reference panel consisting of all individuals from the 1000 Genomes Project (March 2012 release) using IMPUTE2 v2.2.2 (accessed February 2, 2016) and standard procedures with a cutoff of R2 > 0.3. The final number of SNPs following imputation was 24,815,139. The quality of the imputed data was previously validated. 7
UK Biobank Cohort
The UK Biobank consists of approximately 500,000 participants with a wide variety of phenotypic and genotypic information. 8 Ethics approval for the UK Biobank study was obtained from the North West Centre for Research Ethics Committee (11/NW/0382). 8 Genotype data were obtained from the v3 release of UK Biobank. 8 The UK Biobank electronic healthcare records were available for 438,670 individuals of European ancestry and included data until June 2019. Genotype data were imputed centrally by UK Biobank with IMPUTE2 using the Haplotype Reference Consortium and the UK10k+ 1000GP3 reference panels. 9 Metrics for quality control were established and then used to filter DNA variants by UK Biobank. 8 Imputed SNPs were excluded if they had an IMPUTE2 info score <0.4. The final number of SNPs following imputation was 17,136,336.
Database Quality Control
For both the KPRB and UK Biobank cohorts, individuals were excluded if they were outliers based on genotyping missingness rate or heterogeneity, whose sex inferred from the genotypes did not match their self-reported sex, who withdrew from participation, or who were not of European ancestry. The purpose of restricting individuals to those with European ancestry is to reduce population stratification in the study; for example, if the risk of chondromalacia among individuals with African ancestry is higher than that for European individuals, then any SNP with an allele frequency that is different between African and European ancestries would appear to be associated with chondromalacia. Overall, these filters resulted in excluding 18.9% and 3.1% of individuals (mostly due to the ancestry filter) in the KPRB and UK Biobank cohorts, respectively. Genetic variants were excluded that failed quality control procedures in any of the genotyping batches, that showed a departure from Hardy-Weinberg of P < 10−50 or that had a minor allele frequency < 0.004.
Phenotype Definitions
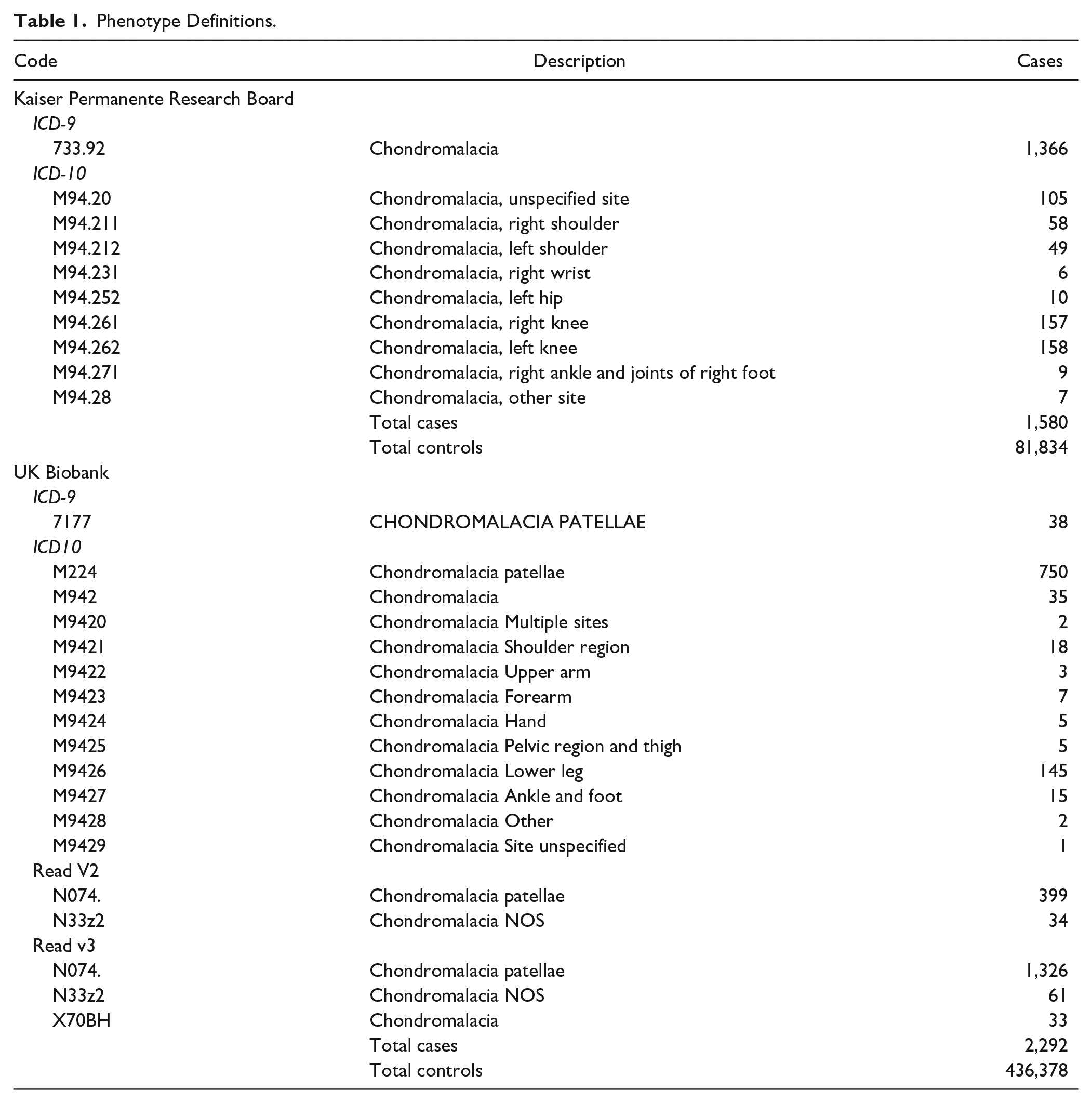
In the KPRB cohort, chondromalacia cases were identified based on clinical diagnoses captured in the Kaiser Permanente Northern California electronic health record system from 1995 to July 22, 2015 ( Table 1 ). International Classification of Diseases, Ninth Revision (ICD-9) or International Classification of Diseases, Tenth Revision (ICD-10) codes were used to identify cases of chondromalacia. In the UK Biobank cohort, chondromalacia cases were also identified from ICD-9 and ICD-10 codes, as well as primary care data (Read v2 or Read v3) ( Table 1 ). Within the UK healthcare setting, individuals seeking advice or treatment for a health concern normally first meet with a family physician (known as a General Practitioner, or GP) or a nurse (e.g., a Nurse Practitioner) at their local general practice. GPs can refer patients who require more specialized treatment (or further tests) to hospital or other community-based services. Read codes are a coded thesaurus of clinical terms used in primary care since 1985. There are 2 versions: version 2 (Read v2) and version 3 (CTV3 or Read v3). Both provide a standard vocabulary for clinicians to record patient findings and procedures.
Phenotype Definitions.
GWA
GWA studies were conducted using PLINK v2.0a2. 10 SNP associations with chondromalacia were tested with a logistic regression model using allele counts for typed and imputed SNPs. The model was adjusted for genetic sex, age, height, weight, and race/ethnicity using 10 principal components. For UK Biobank, age of enrollment was also included. Covariates were ascertained centrally by either KPRB or UK Biobank. Determination of genetic ancestry was performed by principal component analysis (PCA) computed centrally by either KPRB or UK Biobank, as previously described. 8
To account for inflation due to population stratification, the genomic control parameter (λgc) was calculated (λgc = 1.005 for KPRB; λgc = 0.950 for UK Biobank). Subsequently, P values were adjusted for the genomic control in each population.
Results using odds ratios per allele from each cohort were combined by inverse-variance, fixed-effects meta-analysis using PLINK v2.0a2. Here, meta-analysis refers to a statistical method to combine data from GWAS performed on 2 independent cohorts. A total of 9,161,987 SNPs were present in both GWAS and used in the meta-analysis. A P value of P < 5 × 10−8 was used as a threshold for genome-wide significance.
Further bioinformatics investigations of the top genome-wide significant loci from the GWAS were conducted. Quantile-quantile (QQ) and Manhattan plots were created using the R package qqman. Regional association plots were generated for each locus with LocusZoom (accessed November 21, 2020). 11 The genomic context of each SNP was investigated using RegulomeDB (accessed November 21, 2020) 12 web tools. ChIP seq data from the ENCODE project was used to determine whether SNPs were located within transcription factor binding sites. 13 Summary statistics for all SNPs from the GWAS and the meta-analysis will be available at the NHGRI-EBI Catalog of human GWA studies: https://www.ebi.ac.uk/gwas/ upon acceptance of this article.
Supplemental Searches
To investigate how potential polymorphisms may affect nearby gene activity, the GTEX database was queried. This database provides genetic expression quantitative traits on a global scale by finding DNA variants that are associated with changes in nearby genes from a multitude of tissues and cell lines.
Similarly, to determine whether potential polymorphisms influenced the expression of a nearby gene, we searched the ENCODE project database.12,13 The ENCODE project screens for DNA variants located within transcription factor binding sites on a genome-wide level using Chromatin Immunoprecipitation followed by DNA sequencing, which is a method to sequence the DNA region bound by transcription factors in vivo.
Data Sharing
All data will be openly and publicly available upon publication of this article.
Results
Identification of DNA Variants Associated with Chondromalacia
For KPRB, there were 1,580 cases of chondromalacia and 81,834 controls ( Table 1 ). For UK Biobank, there were 2,292 cases and 436,378 controls ( Table 1 ). The demographics for sex, height and weight for the 2 cohorts are shown in Table 2 . Individuals that were taller and heavier had slightly higher risk of chondromalacia. There was no significant difference in the incidence of chondromalacia between men and women.
Study Demographics.
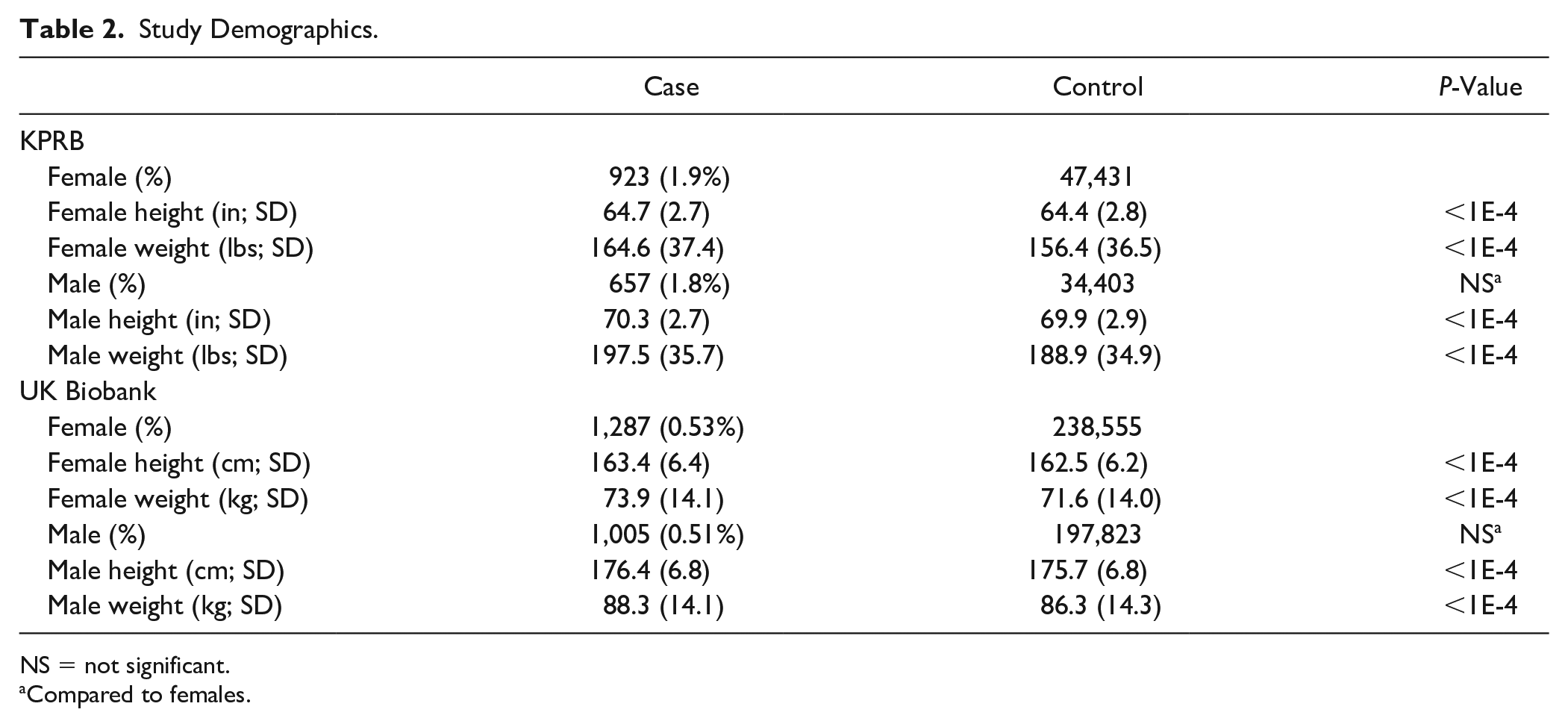
NS = not significant.
Compared to females.
GWA analyses for chondromalacia were performed with the KPRB (83,414 individuals) and UK Biobank (438,670 individuals) cohorts using sex, weight, and height as adjustments (
Table 3
). For UK Biobank, age of enrollment was also included as a covariate. Data from the 2 GWAS were combined in a fixed-effect meta-analysis (
Table 4
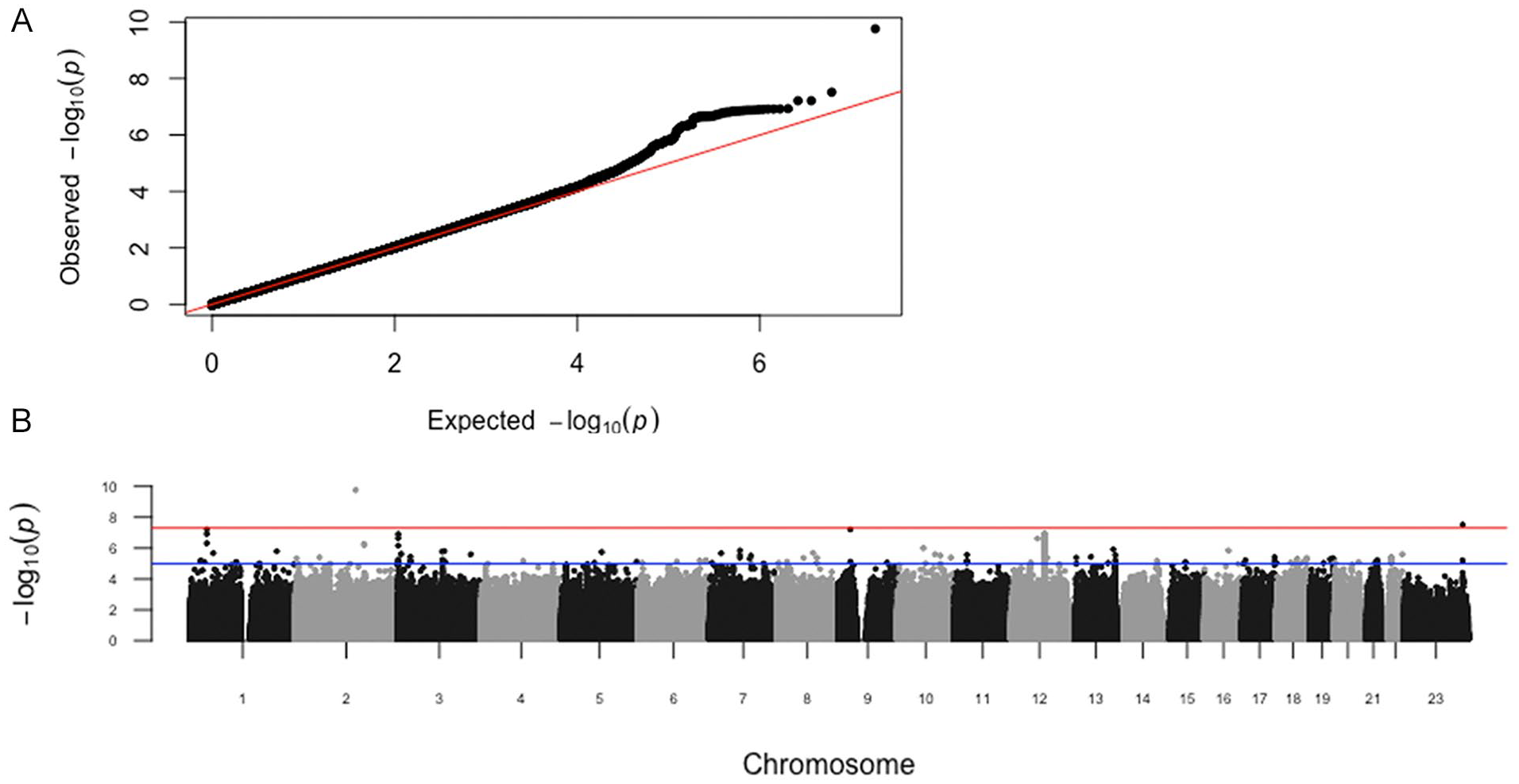
). We compared the observed P values to the distribution of P values expected by chance in a QQ plot (
Summary Statistics.
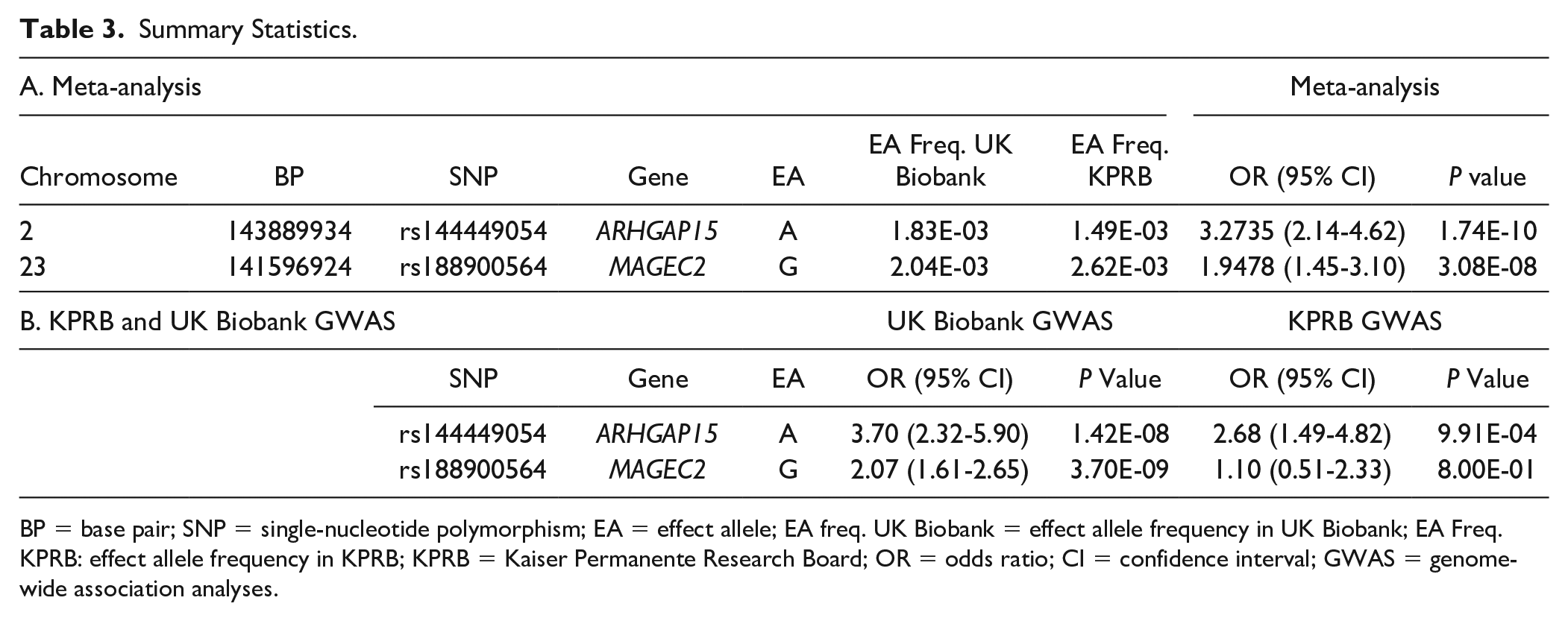
BP = base pair; SNP = single-nucleotide polymorphism; EA = effect allele; EA freq. UK Biobank = effect allele frequency in UK Biobank; EA Freq. KPRB: effect allele frequency in KPRB; KPRB = Kaiser Permanente Research Board; OR = odds ratio; CI = confidence interval; GWAS = genome-wide association analyses.
Genotype Counts for Chondromalacia.
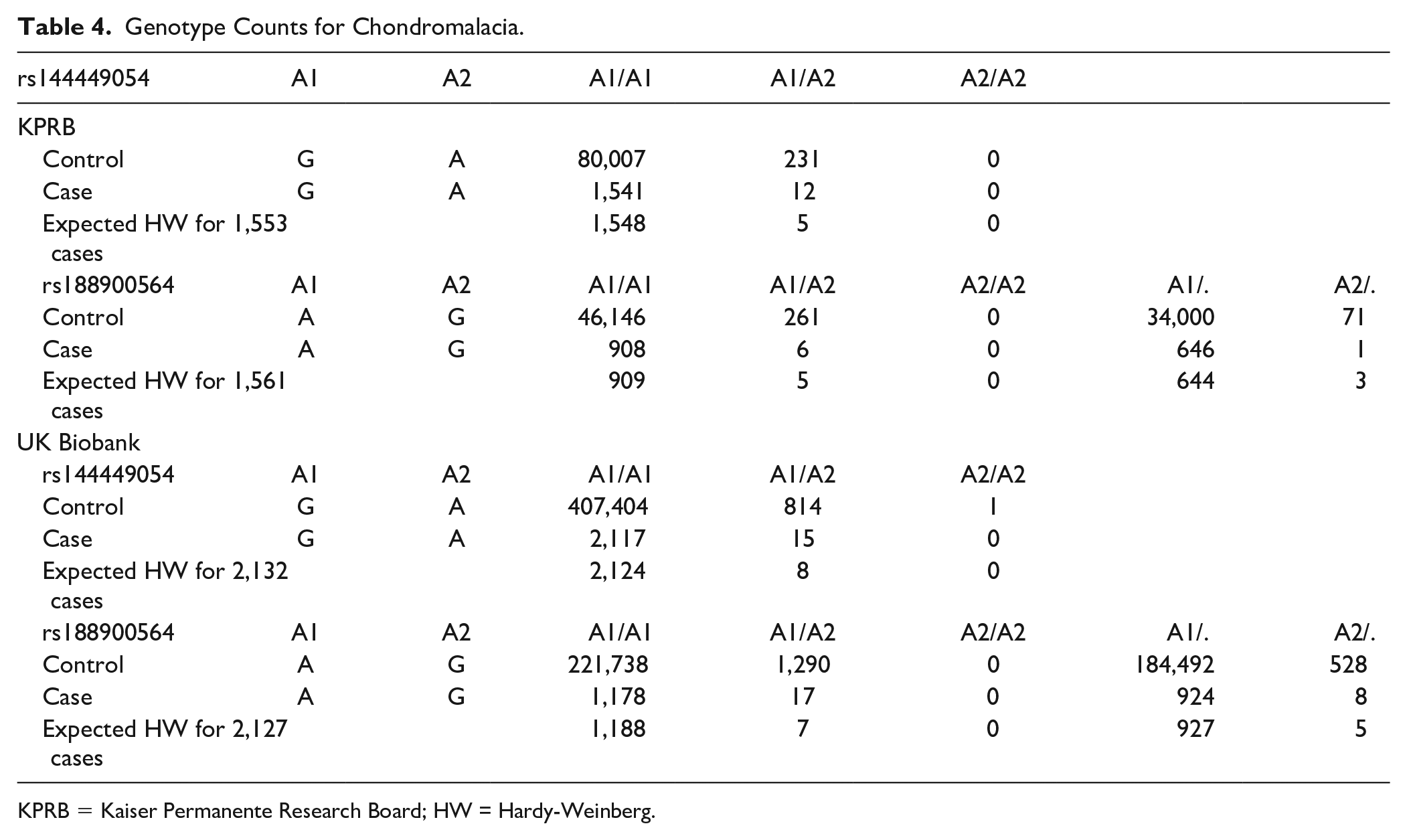
KPRB = Kaiser Permanente Research Board; HW = Hardy-Weinberg.
(
The P value for every SNP from the meta-analysis is shown in a Manhattan plot (
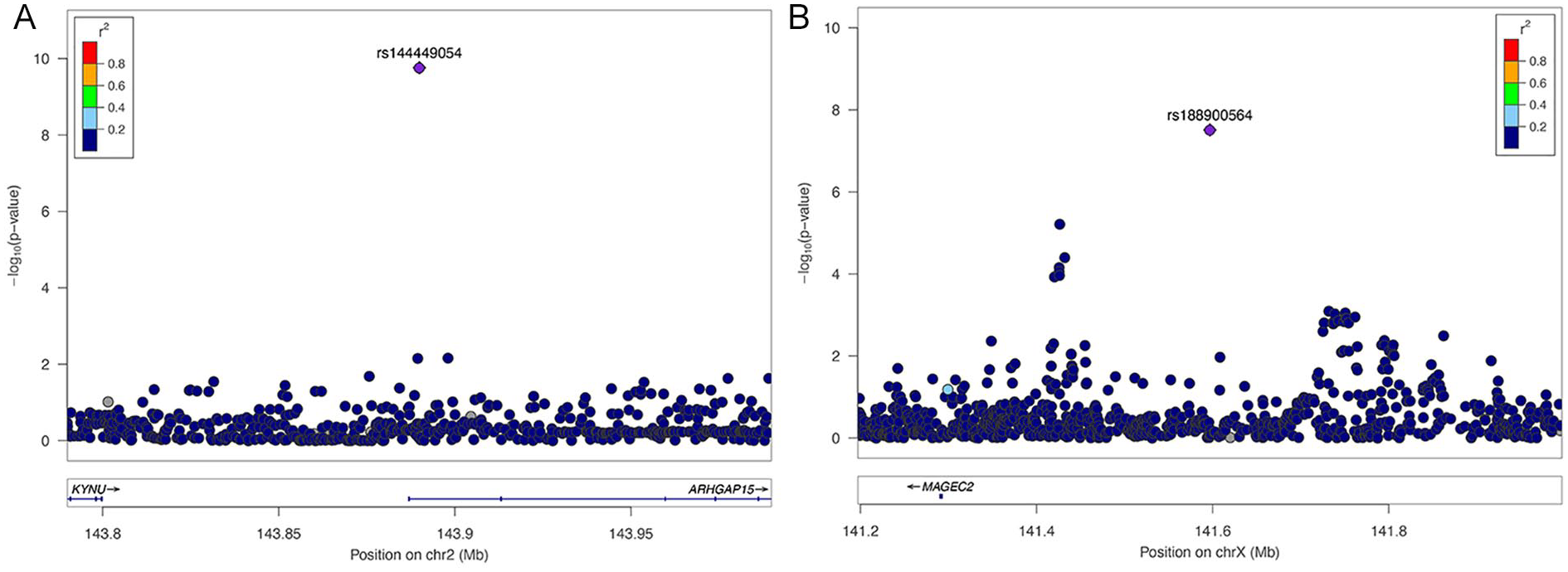
Regional-association plots for chondromalacia. Tested SNPs are arranged by genomic position around the lead SNP (purple diamond). The y-axis indicates −log10 P values for association with chondromalacia for each SNP. The color of dots of the flanking SNPs indicates their linkage disequilibrium (R2) with the lead SNP as indicated by the heat map color key. (
There were no relevant associations linking the identified chondromalacia SNPs with either expression of a nearby gene (GTEX database) or any transcription factors that bind to the DNA sequence containing either of the SNPs (ENCODE database).
Discussion
Genetic Markers for Chondromalacia
To our knowledge, this is the first genetic study of chondromalacia. Two GWA screens using the KPRB and UK Biobank cohorts were performed, and the data were combined in a meta-analysis. Two SNPs (rs144449054 and rs188900564) were identified with associations with chondromalacia that were genome-wide significant, providing insight regarding genetic mechanisms for incurring chondromalacia.
rs144449054 is located on chromosome 2 in the Rho GTPase Activating Protein 15 (ARHGAP15) gene (
rs144449054 is located in an intron of the Rho GTPase Activating Protein 15 (ARHGAP15) that functions in a signaling pathway involved in cytoskeleton reorganization, cell motility, and cell cycle progression. ARHGAP15 is expressed in B and T cells of the immune system, suggesting a role in mediating inflammation leading to chondromalacia. rs188900564 is in the 5′ region of the Melanoma-Associated Antigen C2 (MAGEC2) gene, which enhances the activity of E3 ubiquitin-protein ligases that mediate protein turnover. Given its known biological role, how MAGEC2 influences chondromalacia is not clear. It is also possible that rs188900564 affects the activity of another gene located nearby on the X chromosome, rather than MAGEC2.
Neither of the SNPs affect the protein-coding capacity of a gene. rs144449054 is located in an intron of ARHGAP15 and rs188900564 is located in the 5′ region of MAGEC2. Besides protein-coding changes, another means for a polymorphism to influence gene function is to affect expression of nearby gene(s). For instance, showing that the polymorphism is associated with variation in expression of a nearby gene (i.e., an expression quantitative trait) would not only provide evidence for an effect on expression but would also identify the target gene. Upon querying expression data from the GTEX database using these 2 SNPs, no data were found linking them to expression of a nearby gene.
Another mechanism to influence expression of a nearby gene is if the DNA variation were to occur within the binding region of a transcription factor, which could alter binding of that transcription factor and affect expression. The ENCODE project screens for DNA variants located within transcription factor binding sites on a genome-wide level using chromatin immunoprecipitation followed by DNA sequencing, which is a method to sequence the DNA region bound by transcription factors in vivo. However, querying ChIP seq data from the ENCODE database failed to find any transcription factors that bind to the DNA sequence containing either of the 2 chondromalacia-associated SNPs.
The overall incidence of chondromalacia was higher in KPRB than the UK Biobank. One possibility is that there is a difference in the underlying population between the Bay Area and the United Kingdom. Another possibility is that the difference reflects variability in how chondromalacia is coded between the healthcare systems in the Bay Area and the United Kingdom. In addition, there was no significant difference in sex among the chondromalacia diagnoses. This is in contrast to other investigations that have identified female sex as a risk factor in not only the development of arthritis, but also increased symptomatology of their osteoarthritis.14,15 Reasons for this may again include demographic differences between the KPRB and UK Biobank databases as compared to other populations.
Individuals harboring risk alleles for rs144449054 (A) in ARHGAP15 or rs188900564 (G) in MAGEC2 have an increased risk for chondromalacia. These individuals are present at a frequency of about 0.3% and 0.4% in the European population, respectively. However, for these rare individuals, the relative risk for chondromalacia for rs144449054 or rs188900564 is about 3- and 4-fold, respectively. Although the genetic association results have not yet been validated in an independent study, the 2 genetic markers could provide key information to athletes about their risk for chondromalacia. Genetic testing could allow them to take extra precautions to avoid injury, affect their choice of participation in a particular sport, and also compel them to seek clinical treatment that they might otherwise ignore. The genetic information could also be used by medical professionals to make more informed decisions regarding chondromalacia diagnosis, risk factors, management of the disease, and return-to-play timelines.
Limitations
Our analysis found only 2 genome-wide significant signals, possibly because chondromalacia may be poorly documented in these cohorts. This type of misclassification error would mostly tend to dilute the strength of any signals, if present. Alternatively, it could be that the heritability of chondromalacia phenotypes is low. Another limitation is that the phenotypes were defined from codes contained in electronic health records, and thus we have no information regarding the clinical scenarios surrounding the event, such as whether patients had prior cases of chondromalacia that were not captured. Along these same lines, we have no clinical information on the location of the chondromalacia in each particular joint in some cases. For example, the ICD-10 codes do not allow us to determine the exact location of chondromalacia within the knee, such as to determine condyles or medial/lateral condyle, or tibial plateau. In addition, the cohort included people regardless of whether or not they participated in a sport. For example, we were unable to discriminate if the cases of chondromalacia identified in this study were related to participation in sports or from other causes. Last, this study only evaluated individuals from the European ancestry group, and the effect of rs144449054 (A) in ARHGAP15 or rs188900564 (G) in MAGEC2 in other ethnicities is unknown.
Future Studies
In the future, it will be important to replicate these gene association results with chondromalacia in independent cohorts, especially for athletes. Additional studies are warranted to begin to illuminate the underlying biological mechanism for the association of variation near ARHGAP15 or MAGEC2 with chondromalacia. The results from these studies may validate whether rs144449054 in ARHGAP15 or rs188900564 in MAGEC2 can be used as diagnostic markers to help predict which athletes harbor a higher risk for chondromalacia.
Conclusion
This study provides the first evidence for genetic associations with chondromalacia. Genome-wide significant associations were found for rs144449054 in ARHGAP15 and rs188900564 in MAGEC2 with chondromalacia. This finding could help identify athletes with increased risk for injury, allowing for preventative measures to be used to avoid injury. The 2 genes provide opportunities for future research to uncover molecular and genetic mechanisms underlying chondromalacia.
Footnotes
Author Note
Work performed at Stanford University.
Acknowledgments and Funding
This work was supported by grants from the NIH (5RO1AG025941 and RC2 AG036607). We are deeply indebted to the UK Biobank for providing access to a rich data source, and to the access team for assistance with using the data (Application 17847; “GWAS for risk for sports injuries”). We thank Erik Ingelsson for sharing the database at Stanford containing UK Biobank genotype data.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: SKK is CEO of AxGen, Inc., a genetic testing company for sports injuries. GDA serves as an advisor for AxGen and holds stock options.
Ethical Approval
This study utilized de-identified data and therefore IRB approval was not required.