
Abstract
Objective
Synovial inflammation influences the progression of osteoarthritis (OA). Herein, we aimed to identify potential biomarkers and analyze transcriptional regulatory-immune mechanism of synovitis in OA using weighted gene coexpression network analysis (WGCNA).
Design
A data set of OA synovium samples (GSE55235) was analyzed based on WGCNA. The most significant module with OA was identified and function annotation of the module was performed, following which the hub genes of the module were identified using Pearson correlation and a protein-protein interaction network was constructed. A transcriptional regulatory network of hub genes was constructed using the TRRUST database. The immune cell infiltration of OA samples was evaluated using the single-sample Gene Set Enrichment Analysis (ssGSEA) method. The hub genes coexpressed in multiple tissues were then screened out using data sets of synovium, cartilage, chondrocyte, subchondral bone, and synovial fluid samples. Finally, transcriptional factors and coexpressed hub genes were validated via experiments.
Results
The turquoise module of GSE55235 was identified via WGCNA. Functional annotation analysis showed that “mineral absorption” and “FoxO signaling pathway” were mostly enriched in the module. JUN, EGR1, FOSB, and KLF4 acted as central nodes in protein-protein interaction network and transcription factors to connect several target genes. “Activated B cell,” “activated CD4T cell,” “eosinophil,” “neutrophil,” and “type 17 T helper cell” showed high immune infiltration, while FOSB, KLF6, and MYBL2 showed significant negative correlation with type 17 T helper cell.
Conclusions
Our results suggest that the expression level of apolipoprotein D (APOD) was correlated with OA. Furthermore, transcriptional regulatory-immune network was constructed, which may contribute to OA therapy.
Keywords
Introduction
Osteoarthritis (OA), the most common musculoskeletal disorder, can lead to disability and reduce the quality of life in the elderly. 1 To date, no available treatment has been effective in preventing OA disease progression, although joint replacement surgery remains an option. 2 A number of local and systematic risk factors, including sports injury, anatomical abnormity, obesity, genetics, aging, and low-grade systemic inflammation contribute to the initiation and development of OA. 3 The pathology of OA involves multiple joint tissues, such as synovium, cartilage, subchondral bone, and synovial fluid. Therefore, OA is considered as a whole joint disease. 4
Synovium inflammation contributes to the development of OA. 5 The histological changes in OA synovium include synovial lining hyperplasia, sublining fibrosis, increased angiogenesis, and immune cells infiltration. 6 Production of proinflammatory cytokines and infiltration of immune cells can aggravate the progression of OA.5,7 Therefore, the OA synovium may release mediators that could be potential biomarkers and therapeutic targets for OA.8,9
In the era of big data, transformation of biomedical big data into valuable knowledge has been one of the most important challenges in bioinformatics. 10 Weighted gene coexpression network analysis (WGCNA) can identify biologically significant modules and genes associated with clinical traits. 11 WGCNA has been applied in varieties of data, including gene expression profiles, genetic marker data, proteomics data, metabolomic data, and other high-dimensional data.12,13
In this study, we analyzed synovitis samples from patients with OA using the WGCNA method to identify significant modules, along with function annotation of module genes and hub genes. Then, a protein-protein interaction (PPI) network and a transcriptional regulatory-immune network of hub genes were constructed; hub genes coexpressed in synovium, cartilage, chondrocyte, subchondral bone, and synovial fluid tissues were then screened out. Finally, transcriptional factors and coexpressed hub genes were validated via quantitative reverse transcription PCR (RT-qPCR) and enzyme-linked immunosorbent assay (ELISA).
Materials and Methods
Data Collection from Patients with OA and Control Donors
Data sets were downloaded from the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/), including GSE55235, GSE55457, GSE82107, GSE89408, GSE1919, GSE114007, GSE57218, GSE110606, and GSE51588. GSE55235, GSE55457, and GSE1919 data sets include OA synovial tissue samples, normal synovial tissue samples, and rheumatoid arthritis (RA) synovial tissue samples. GSE89408 includes OA, normal, RA, and other lesions synovial tissue samples. GSE82107 includes OA and normal synovial tissue samples. OA and normal synovial tissue samples were used for further research.
GSE114007, GSE129147, GSE57218, and GSE110606 data sets include OA and normal cartilage or chondrocyte samples. Another data set of cartilage samples, “Ann Rheum Dis,” was downloaded from the supplementary materials of an original article. 14 GSE51588 includes OA and normal subchondral bone samples. Each sample has lateral and medial tibial plateaus. We divided this group into two data sets and named them as “OA-ND” and “MT-LT.” The OA-ND data set includes OA and normal subchondral bone samples. The MT-LT data set includes OA medial samples and relatively normal lateral samples from OA patients. The proteomics data set of synovial fluid, “Arthritis Rheum” was integrated from proteomic analysis of synovial fluid. 15 The details for all data sets are shown in Supplementary Material S1.
Data Preprocessing and Identification of Differentially Expressed Genes (DEGs)
The GEO data set files were downloaded and processed by the GEOquery package using R language (version 3.6.3, http://r-project.org/). 16 The probes corresponding to multiple genes were deleted. Considering the existence of one gene corresponding to multiple probes, the average expression of multiple probes was taken as the expression value of the gene. Differentially expressed genes (DEGs) were analyzed using “limma” or “edgeR” package for all data sets, according to the gene expression data.17,18 The cutoff criteria of DEGs was |fold change (FC)|>2 and P < 0.05. Data filtering for differentially proteomic analysis of synovial fluid was performed using |FC|≥1.2 and P < 0.05. The results of GSE110606, “Ann Rheum Dis,” and “Arthritis Rheum” were directly downloaded from Supplementary Materials.14,15,19 The details for all data sets preprocessing are shown in Supplementary Material S2.
Construction of Coexpression Modules Associated with OA Based on WGCNA
The R package WGCNA was used to construct a coexpression network. First, samples of synovium data set GSE55235 were clustered to detect and eliminate outliers using the flashClust function. 20 The independence and average connectivity degrees of the coexpression networks were mainly determined by a power value. 21 Then, suitable soft thresholding power value was selected using the pickSoftThreshold function of the WGCNA algorithm if the independence degree was >0.85. 22 Then, a hierarchical clustering tree was generated with the power value with a module minimum size cutoff of 30. 23 Genes with similar expression data were clustered and formed a branch of the tree, representing a gene module.
Identification of Clinically Significant Modules and Functional Annotation
In this process, gene significance (GS), module eigengene (ME), and module membership were calculated. GS was defined as the absolute value of the correlation between a gene and a trait. ME was the first principal component of the given module. Module membership was defined as the correlation between ME and the gene expression profile. The significance between ME and a clinical trait was measured using Pearson correlation. We selected the module of highest correlation coefficient with OA trait. 11 Functional annotation analysis of module genes, including gene ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes pathways (KEGG), was conducted using the online tool DAVID6.8 (https://david.ncifcrf.gov/). GO terms contain biological process, cell component, and molecular function. P < 0.05 was considered as the threshold, and the top seven significant results were visualized using R.
Identification of Hub Genes and Construction of PPI Network
Hub genes are defined as genes that have high connectivity inside coexpression modules. The hub genes of the significant module were determined through an absolute value of the gene module membership >0.7. Furthermore, all hub genes were mapped into the Search Tool for the Retrieval of Interacting Genes (STRING) database (v11.0; https://string-db.org/). A combined score of >0.4 was considered significant. The PPI network was visualized using Cytoscape software (version 3.7.1), and the top 20 hub genes were calculated using the cytoHubba plugin through the maximal clique centrality method. 24
Construction of Transcriptional Regulatory Network of Hub Genes and Calculation of Immune Cell Infiltration Score in OA
First, we downloaded 8,472 regulatory relationships between human transcription factors and target genes from TRRUST (v2) database to construct a background transcriptional regulatory network, and all hub genes of synovium OA data set GSE 55235 were mapped into this network. 25 We extracted the one-step neighborhoods of the hub nodes to build a transcriptional regulatory network of hub genes. A list of transcription factors was obtained from AnimalTFDB, including 1,557 known human transcription factors. 26 Cytoscape software was used to network visualization in which hub nodes, transcription factors, and target genes were marked in different ways in the network. Then, we got a metagene that contained 782 genes presenting 28 immune cell types (Suppl. Material S3). 27 The immune cell infiltration score of 28 immune cell types of GSE55235 synovium samples was determined using single-sample Gene Set Enrichment Analysis (ssGSEA) software implemented in the R GSVA package.27,28
Construction of Transcription Factor and Immune Cell Network
The transcription factors in the hub genes transcriptional regulatory network were selected. We calculated the Pearson correlation coefficient between the expression of transcription factors and the infiltration score of immune cell types. The significant correlation between transcription factors and cell types was retained to construct a network (P < 0.01, |cor| > 0.6), which was displayed using Cytoscape visualization tool. The transcription factors and cells were displayed with different node shapes, and the correlation between them was expressed using different-colored lines.
Screening of Coexpressed Hub Genes in Multiple Tissues
DEGs and differentially expressed proteins of synovium, cartilage, chondrocyte, subchondral bone, and synovial fluid tissues were selected for hub genes validation. First, we obtained intersections of synovium GEO data sets, including GSE55457, GSE82107, GSE89408, and GSE1919. Second, intersections of cartilage and chondrocyte samples were presented, including GSE114007, GSE57218, “Ann Rheum Dis,” GSE129147, and GSE110606. Moreover, we integrated intersections of GSE51588 (OA-ND), GSE51588(MT-LT), and “Arthritis Rheum.” Data analysis and visualization were produced using an online tool (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Validation of Transcription Factors and Coexpressed Hub Genes with RT-qPCR and ELISA Experiments
To further validate the expression of transcription factors and coexpressed hub genes, articular cartilage, synovium, subchondral bone, and synovial fluid of OA samples were obtained at the time of total joint arthroplasty (age 53-70 years, Kellgren-Lawrence 4 grade). Control samples of synovial fluid were collected from patients receiving arthroscopic surgery, including meniscectomy and free body removal (age 42-54 years, Kellgren-Lawrence 0-1 grade). This study was approved by the institutional review board, and informed consent was obtained from each donor.
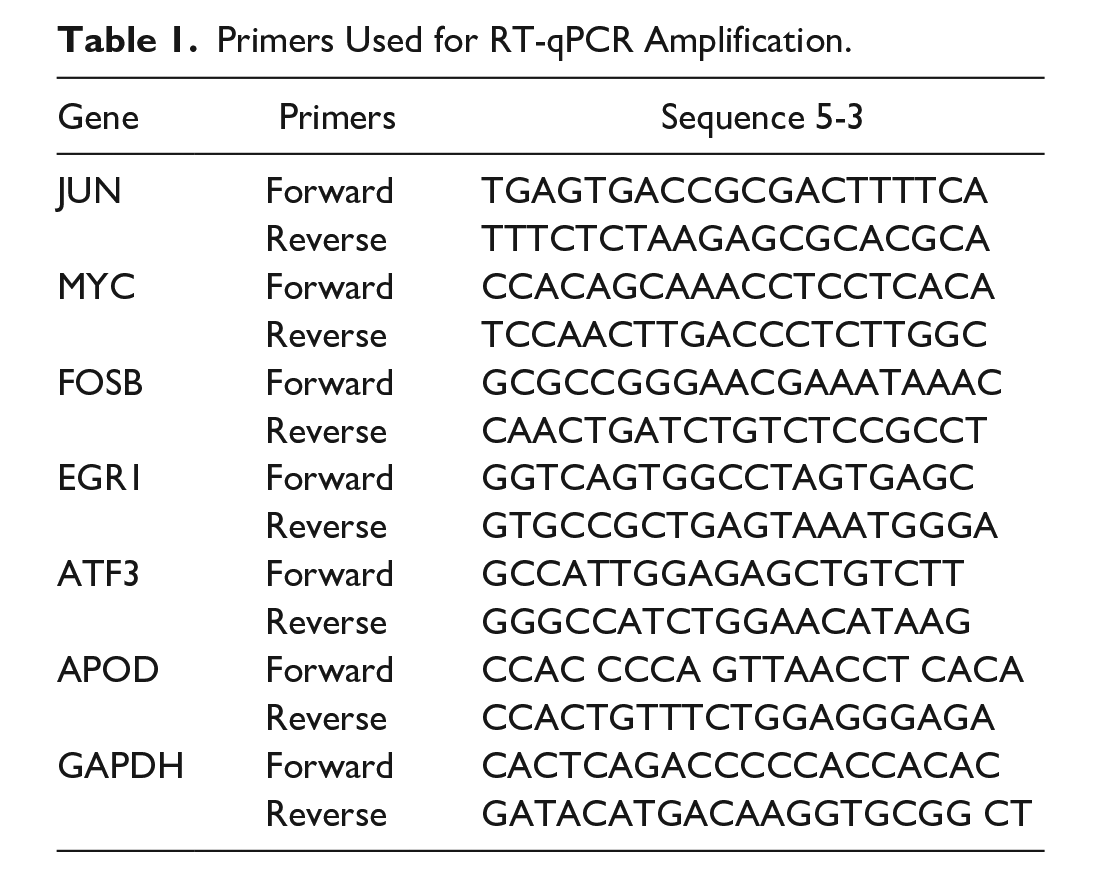
Isolation of fibroblast-like synoviocyte (FLS) and chondrocyte was conducted as previously reported.29,30 Passage 8 of FLS and passage 2 of chondrocyte were used for further experiments. 31 FLS and chondrocytes were seeded in 6-well plates with or without stimulation with 10-ng/mL interleukin-1β (IL-1β) for 48 hours. Medium supernatant, subchondral bone samples, and synovial fluid samples were preserved in liquid nitrogen until being utilized. RNA isolation, reverse transcription cDNA synthesis, and quantitative PCR (RT-qPCR) were undertaken following protocols (ESscience Biotech, Shanghai, China; Bio-Rad, Hercules, CA). The primer sequences of transcription factors and coexpressed hub genes were listed in Table 1. The data were analyzed using 2-ΔΔCq method and GAPDH served as internal controls. ELISA was performed to confirm the protein expression of coexpressed hub genes in medium supernatant and synovial fluid samples (Elabscience Biotech, Wuhan, China; Tecan, Switzerland). All experiments repeated at least three times.
Primers Used for RT-qPCR Amplification.
Statistical Analysis
The results are presented as mean ± the standard error of the mean (SEM). Statistical analysis was performed using GraphPad Prism 7.0 software (GraphPad Software Inc., San Diego, CA). The differences between groups were determined using the Student t test. P < 0.05 was considered statistically significant.
Results
Results of Data Processing
An overall flow chart of the study design is presented in Figure 1 . The synovium data set GSE55235 of OA was examined using DEG analysis, WGCNA analysis, function annotation, identification of hub genes, PPI interaction, and immune infiltration. The hub genes were screened with data sets of multiple tissues, while transcription factors and coexpressed hub genes were validated with RT-qPCR and ELISA experiments. A total of 919 genes were differentially expressed in the data set of GSE55235, including 454 upregulated genes and 465 downregulated genes (Suppl. Material S4). Findings related to the DEGs are presented as a heatmap and volcano plot ( Fig. 2 ). For GSE110606 chondrocyte samples, DEGs were directly downloaded from the supplementary materials of an original article. 19 For the GSE51588 data set, DEGs were selected from OA-ND and MT-LT subchondral bone samples. For proteomics analysis, a final list of 63 proteins that were differentially expressed in late OA patients were identified, excluding the proteins that were inconsistent across the two experiment sets. 15 For all data sets, the DEGs and the proteomics analysis details are shown in Supplementary Material S2.
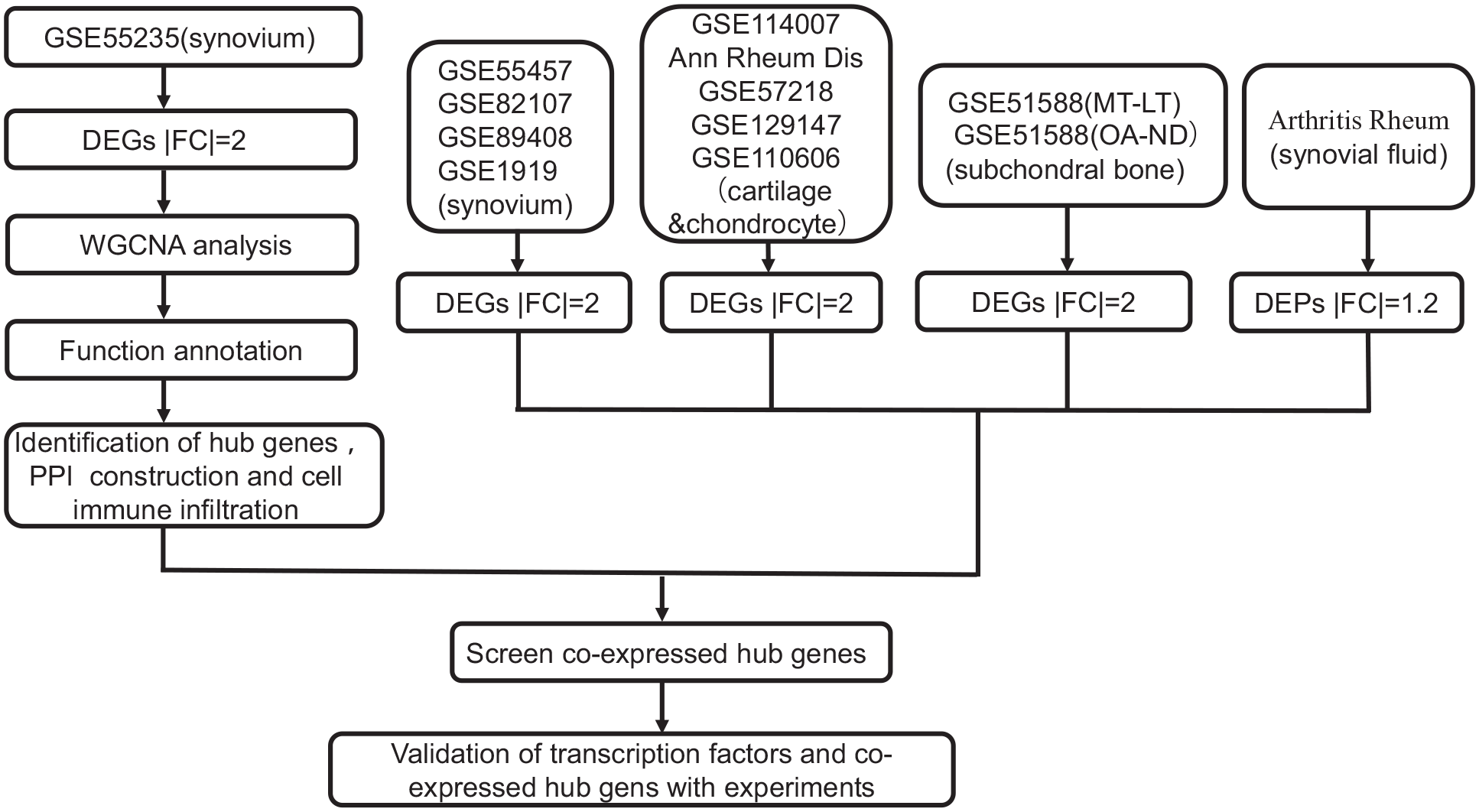
Flow diagram of the study design. The synovium data set GSE55235 of osteoarthritis was analyzed using differentially expressed genes analysis, weighted gene coexpression network analysis, function annotation, identification of hub genes, protein-protein interaction network analysis, and step-by-step immune infiltration. Then, the hub genes were screened with data sets of multiple tissues. Finally, transcriptional factors and coexpressed hub genes in multiple tissues were validated with experiments. WGCNA = weighted gene coexpression network analysis; PPI = protein-protein interaction.
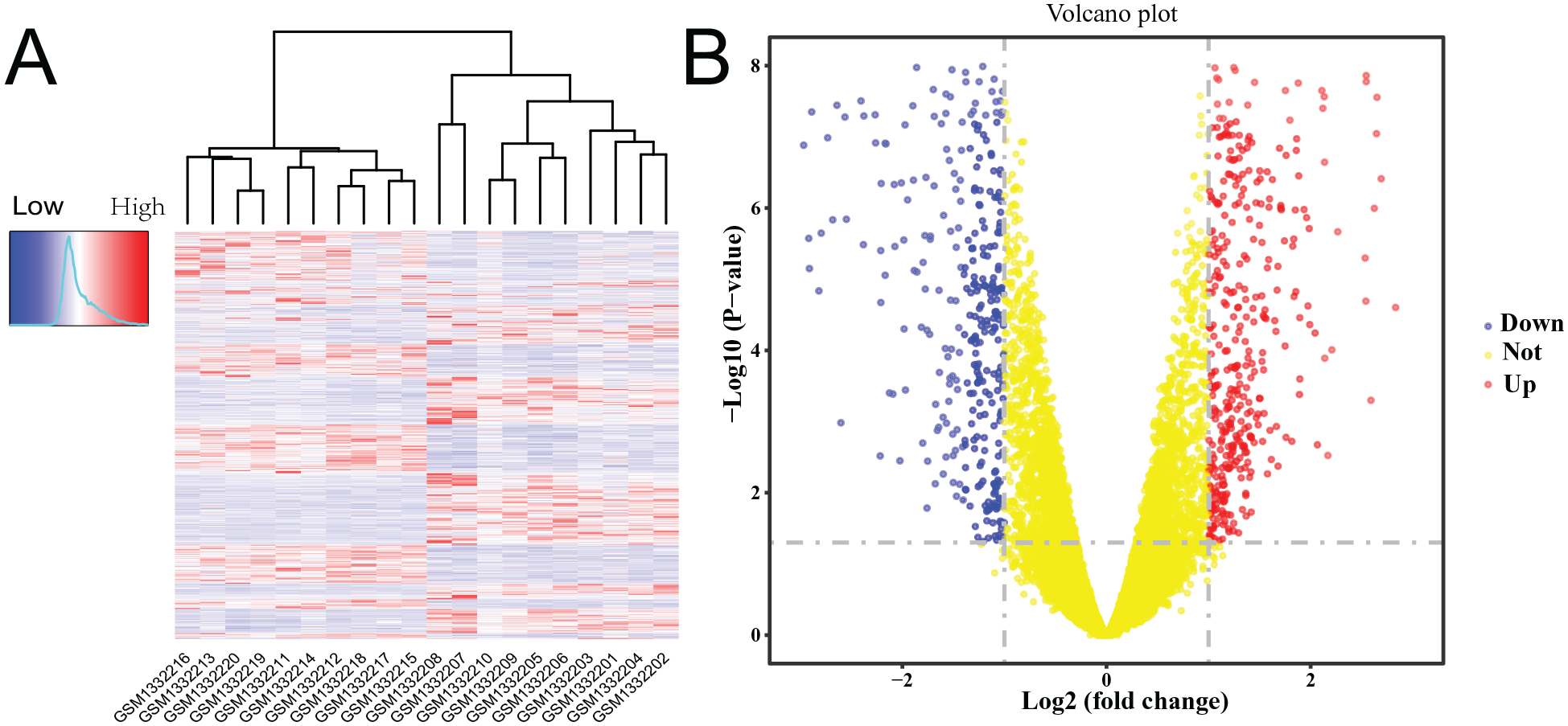
Screening of differentially expressed genes (DEGs) from the OA synovium data set GSE55235.
Construction of Modules Associated with OA Based on WGCNA
For sample clustering analysis, no outlier samples were found using the flashClust function in the synovium data set GSE55235 (
Fig. 3A
). Gene coexpression modules were established for the expression of DEGs using the WGCNA method. An experienced soft threshold value of 6, which was the lowest power value for an independence degree of up to 0.85, was selected to construct a scale-free network (
Fig. 3B
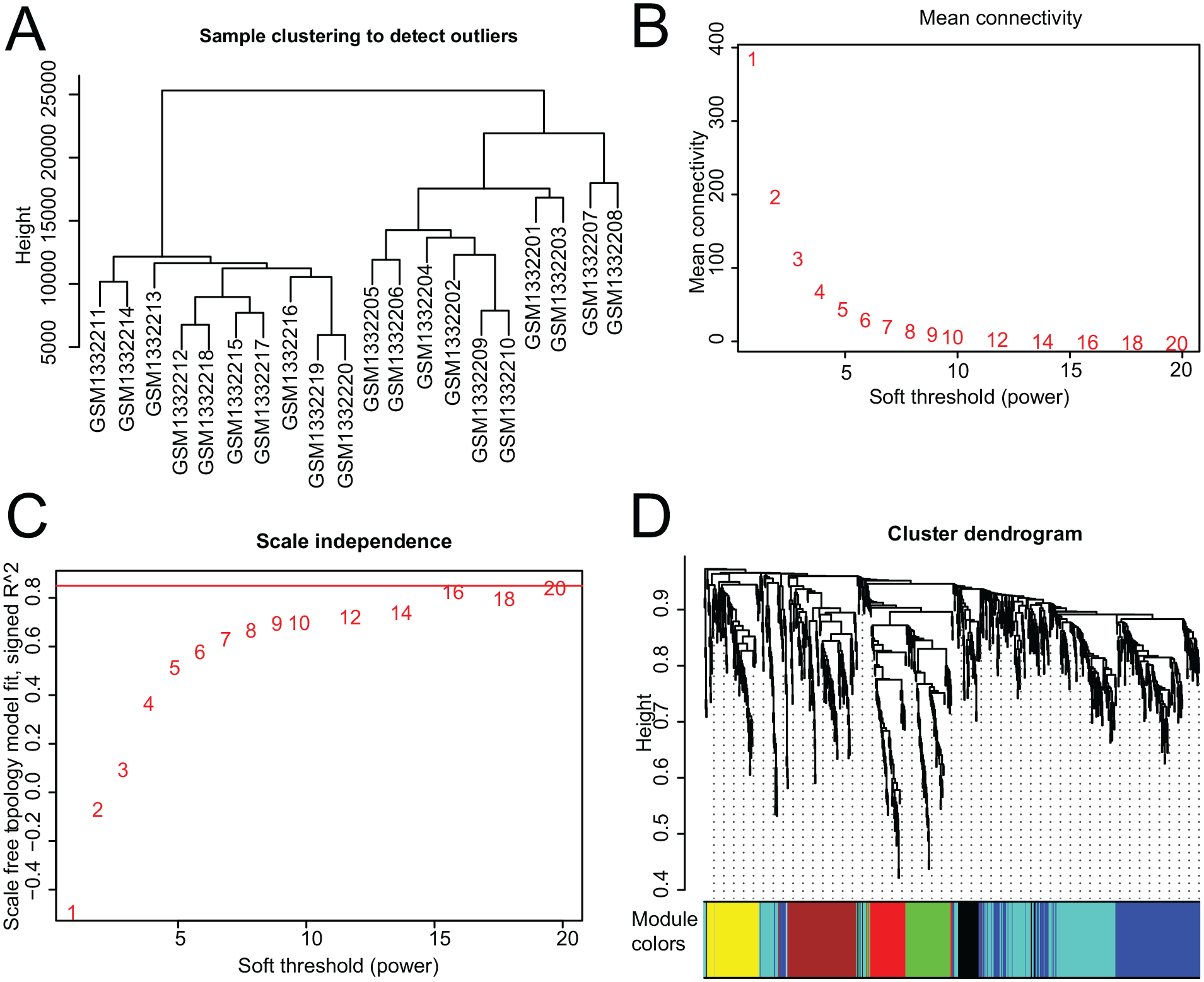
Construction of modules based on weighted gene coexpression network analysis.
Identification of the Most Significant Modules Associated with OA
A heatmap was drawn based on the interactive relationships of seven modules with the OA traits. The turquoise module was significantly correlated to the disease status (Cor = 0.95, P = 2e−10) (
Fig. 4A
). Module and OA status clustering showed that the turquoise module was the most correlated with OA trait (
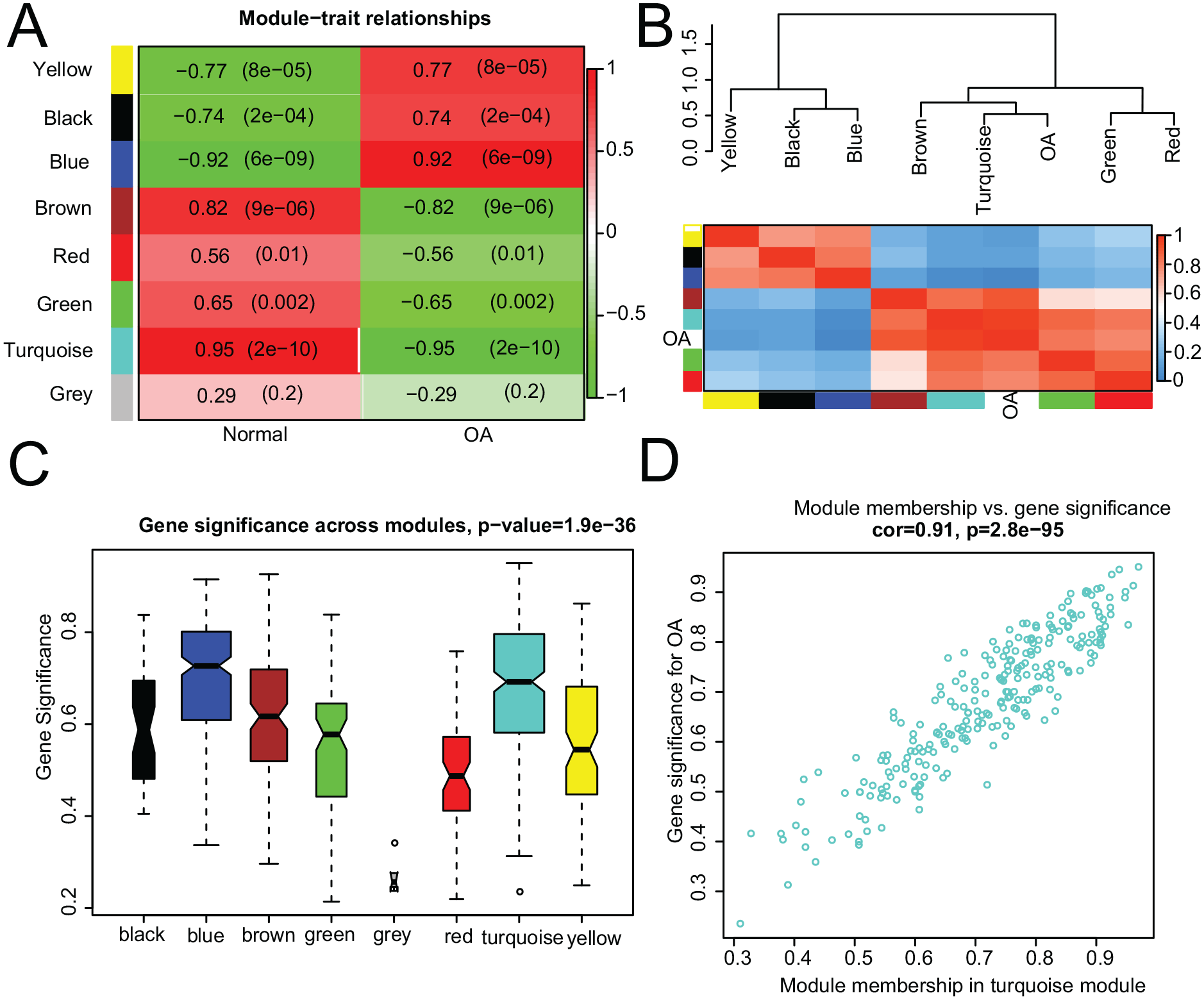
The combination of heatmap
Functional Annotation Results of Module Genes
To further explore the functions of turquoise module, the online tool DAVID was used to investigate the GO terms and KEGG pathways. The biological processes of the turquoise module were mainly enriched in cellular response to zinc ion, negative regulation of growth, cellular response to interleukin-1, nucleus, transcription factor activity, and RNA polymerase II core promoter proximal region sequence-specific binding ( Fig. 5A-C ). The KEGG pathway analysis revealed that the turquoise module was mostly enriched in mineral absorption, FoxO signaling pathway, MAPK signaling pathway, tumor necrosis factor (TNF) signaling pathway, osteoclast differentiation, and cytokine-cytokine receptor interaction ( Fig. 5D ). Metallothioneins were involved in the mineral absorption pathway (Suppl. Material S6).
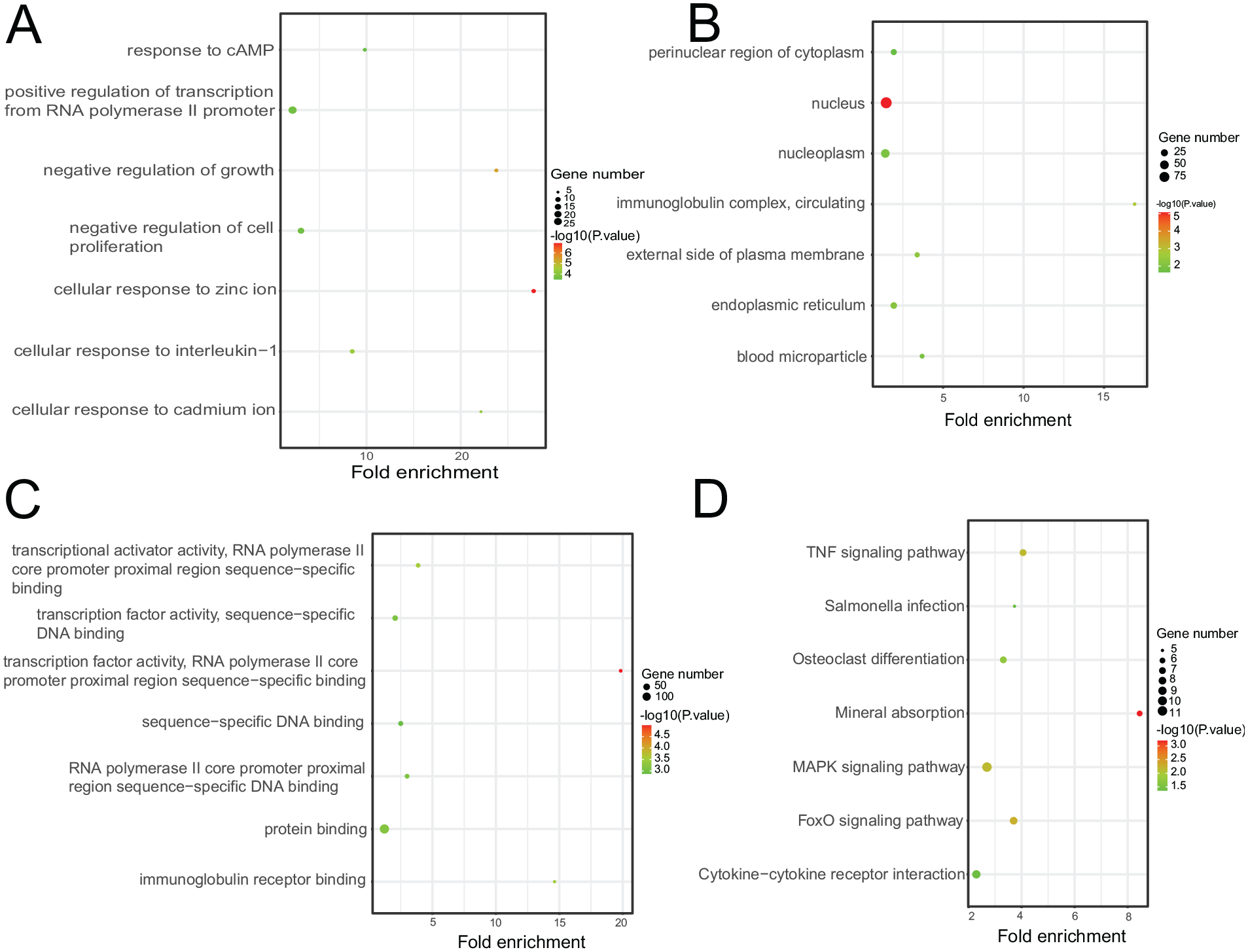
Function annotation of the turquoise module genes showing
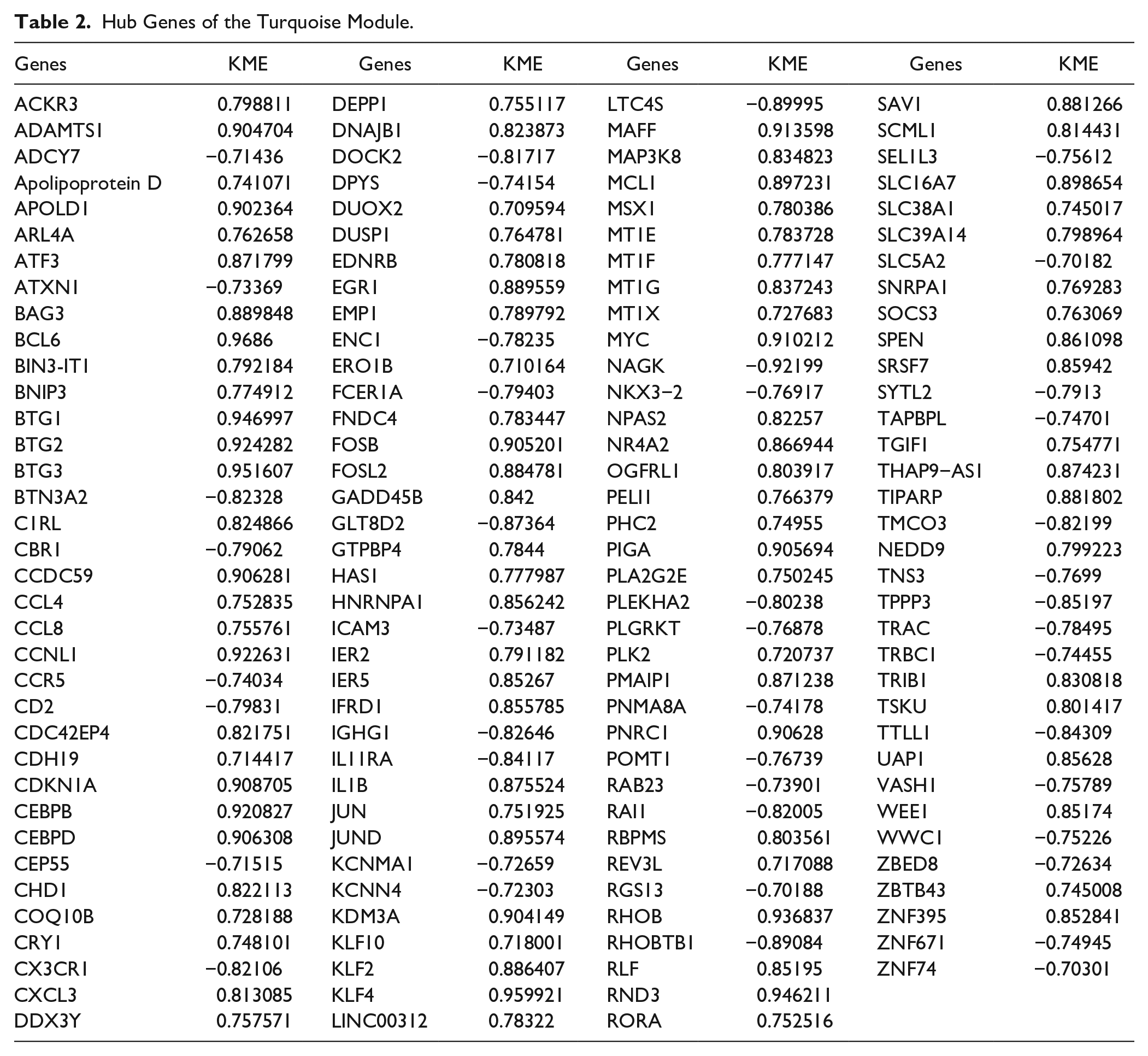
Identification of Hub Genes and Construction of the PPI Network
Genes with high connectivity inside coexpression modules were considered as hub genes. A total of 143 hub genes in the turquoise module were identified with a module membership of >0.7. All hub genes are shown in Table 2 . To further investigate the PPIs among these hub genes, a PPI network was constructed using the STRING database. A total of 251 interactions were included with a combined score of >0.4 ( Fig. 6A ). The top 20 genes based on the maximal clique centrality method included JUN, ATF3, DUSP1, EGR, CDKN1A, FOSB, and MYC ( Fig. 6B ).
Hub Genes of the Turquoise Module.
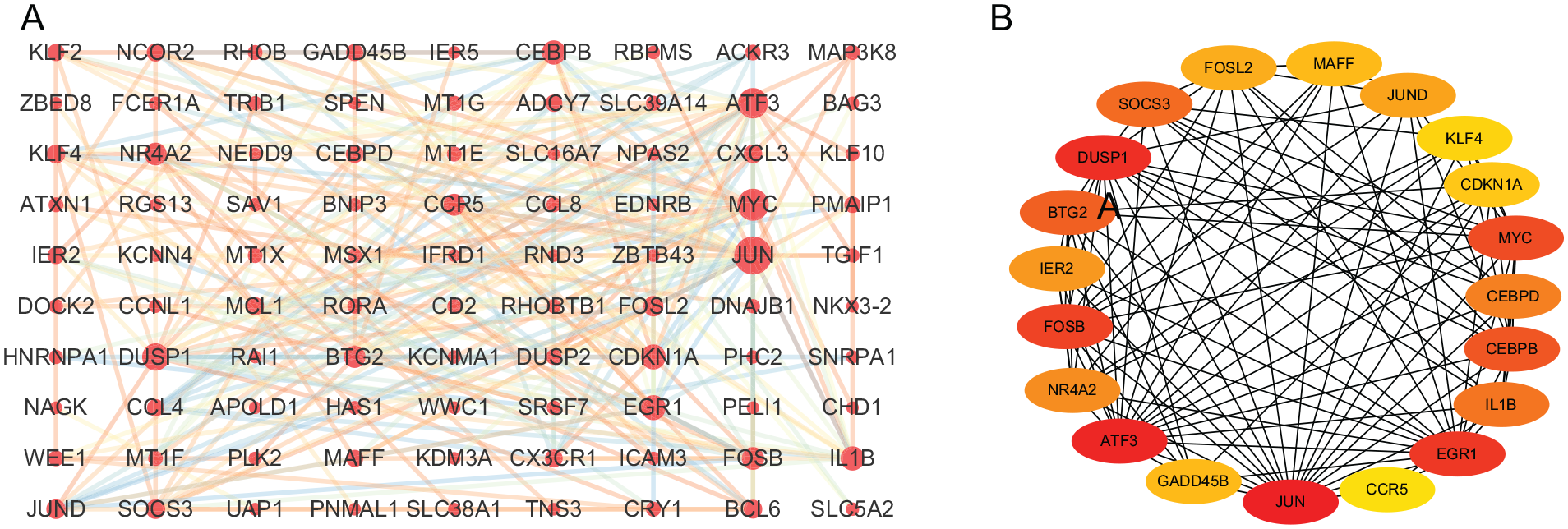
Protein-protein interaction network construction of hub genes in the turquoise module. (
Transcriptional Regulation and Immune Characterization of Hub Genes
A total of 8,472 relationships between transcription factors and target genes were downloaded from TRRUST database, which contained 796 transcription factors and 2,492 target genes, while 90 hub genes were mapped to the TRRUST transcriptional regulatory networks. In addition, one-step neighbourhoods of hub genes were selected to construct a transcriptional regulatory network of hub genes. Finally, this subnetwork contained 3,110 edges and 649 nodes (Suppl. Material S7-S10). In the network, hub genes such as JUN, EGR1, JUND, KLF4, and MYC acted as central nodes and transcription factors to connect a large number of target genes. The biggest degree node, SP1, regulated by EGR1, also regulated the highest number of target genes in the network as a transcription factor
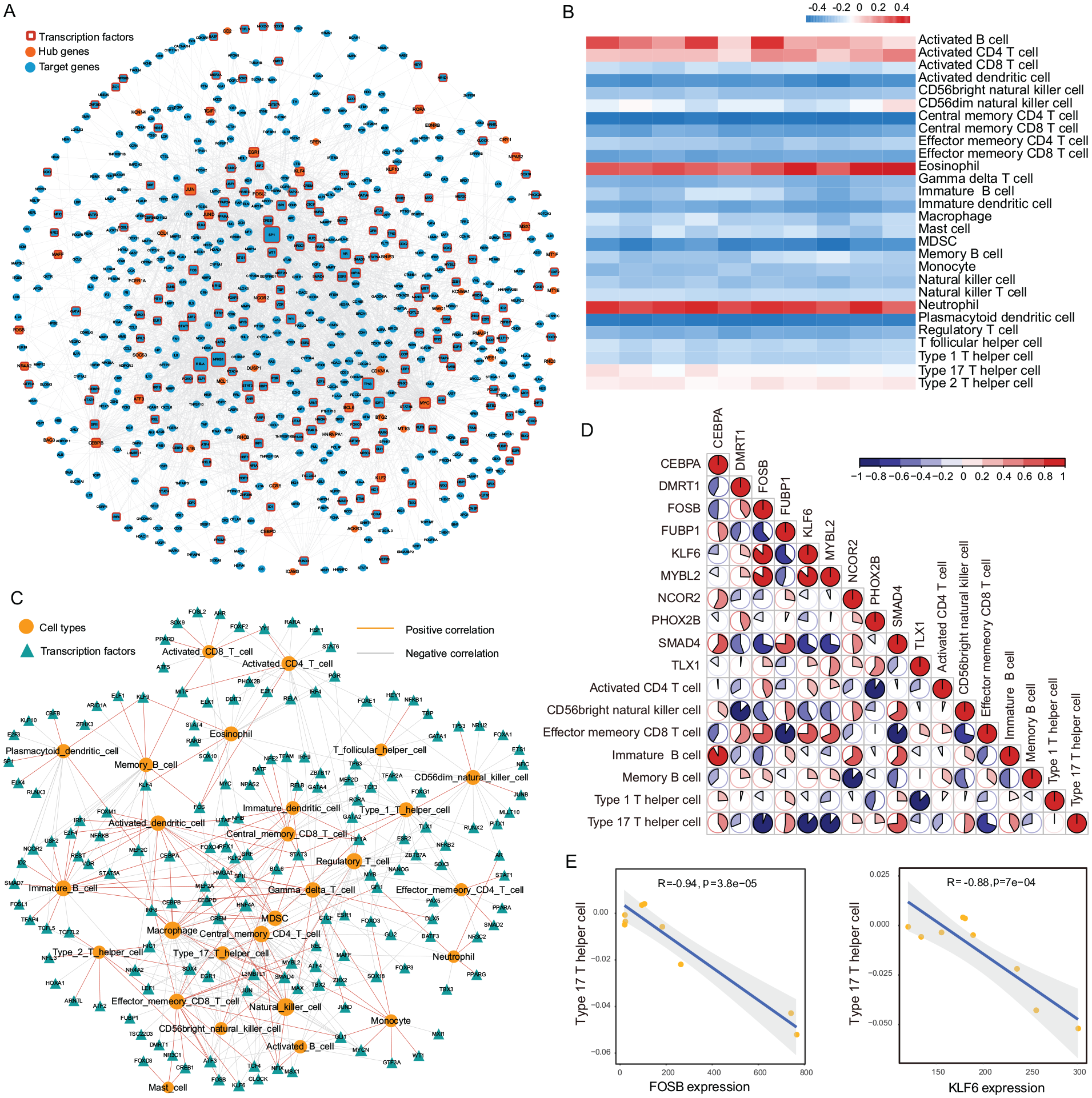
Transcriptional regulation and immune characterization of hub genes.
Screening of Coexpressed Hub Genes in Multiple Tissues
A total of 12 data sets were used for validation of hub genes, including synovium sample data sets of GSE55457, GSE82107, GSE89408, and GSE1919; cartilage/chondrocyte sample data sets of “Ann Rheum Dis,” GSE114007, GSE129147, GSE57218, and GSE110606; subchondral bone sample data sets of GSE51588 (OA-ND) and GSE51588 (MT-LT); and synovial fluid sample data set of “Arthritis Rheum.” A Venn diagram showed that APOD was the only gene in synovium, synovial fluid, and subchondral bone tissues (
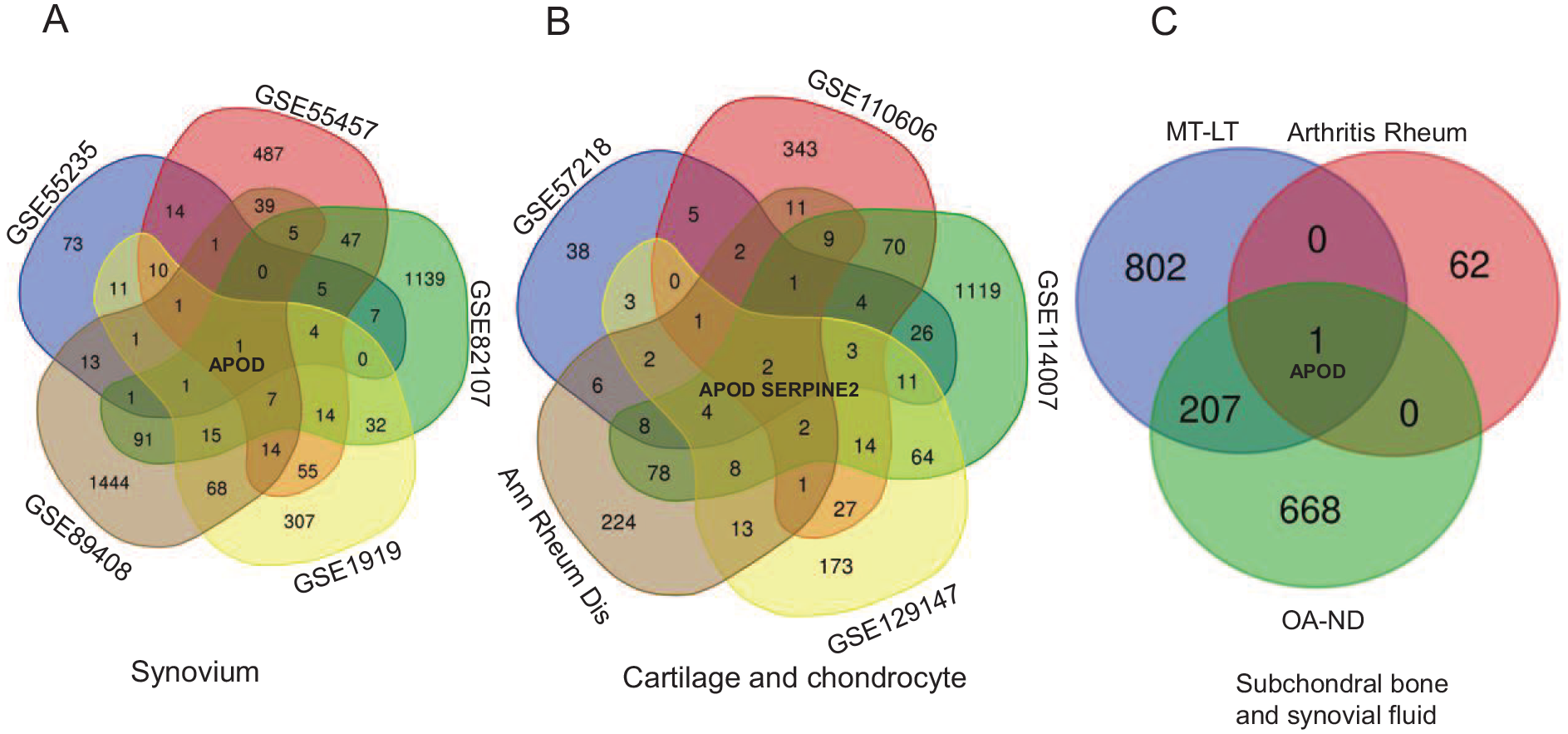
Screening coexpressed genes with multiple tissue data sets.
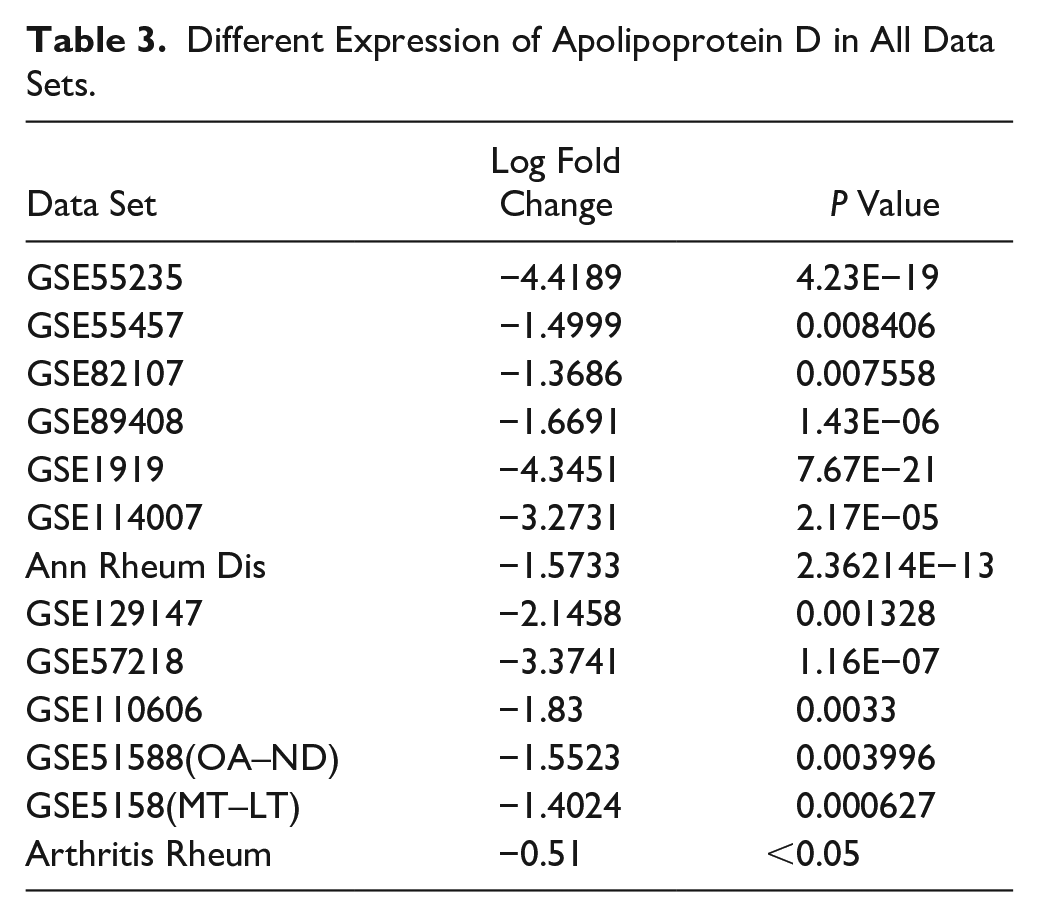
Different Expression of Apolipoprotein D in All Data Sets.
Validation of Transcription Factors and Coexpressed Hub Genes
Transcription factors such as JUN, MYC, FOSB, EGR1, and ATF3 were validated using RT-qPCR. The expression of APOD was identified using RT-qPCR and ELISA in different tissues. We isolated RNA from relatively normal lateral tibia and sclerosis area of the medial tibia (Fig. 9E). Results showed that the mRNA expression of JUN, FOSB, and EGR1 was significantly decreased in IL-1β stimulated FLS (Fig. 9A). The mRNA expression of APOD was downregulated in FLS, chondrocyte, and subchondral bone samples with OA (Fig. 9B-D). The protein expression of APOD in medium supernatant of IL-1β stimulated FLS and chondrocyte and OA synovial fluid was also downregulated (
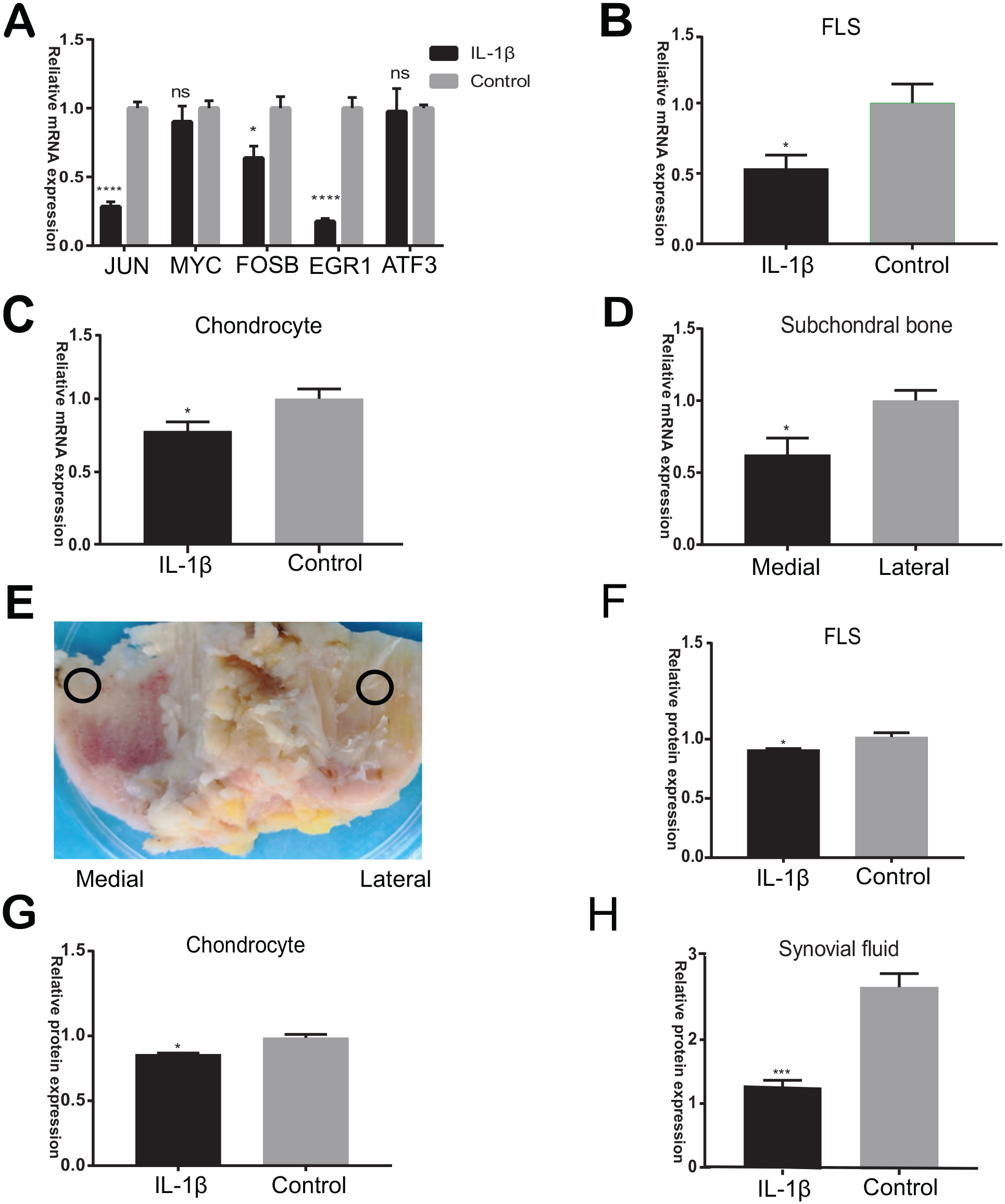
Validation of transcription factors and APOD expression.
Discussion
Abnormal synovial changes can occur at different stages of OA. 32 Inflammation of the synovial membrane can produce proinflammatory and catabolic mediators such as cytokines, prostaglandin E2, nitric oxide, and neuropeptides, which can accelerate the breakdown of cartilage. 9 Indeed, an increasing number of studies have indicated that immune infiltration plays important roles in OA progression. 33 The synovial membranes of OA patients often exhibit immune infiltration, such as in macrophages, T cells, mast cells, B cells, plasma cells, natural killer cells, dendritic cells, and granulocytes. 34 Based on the WGCNA method for synovium tissue transcription analysis, the most significant module associated with the OA trait was identified and APOD was identified a potential biomarker for OA diagnosis. Furthermore, transcriptional factors including JUN, MYC, EGR1, JUND, KLF4, FOSB, and CEBPB were identified, and a transcriptional regulatory-immune network was constructed.
In the OA synovium data set, the turquoise module was the most significant and the correlation value was 0.95. The KEGG pathways of the module were mostly enriched in mineral absorption, FoxO signaling pathway, and MAPK signaling pathway. Metallothioneins (MT1M, MT1F, MT1G, MT1H, MT1X, and MT1E) are involved in the mineral absorption pathway. Evidence has shown metallothioneins play critical roles in immunity, infections, protection against oxidative stress, and regulating immune cells,35,36 and may have potential in the treatment of OA. FoxO can promote cell cycle arrest, DNA damage repair, autophagy, and scavenging of free radicals, and protect cells against various stressors such as oxidative stress. 37 Expression and activation of FoxO play important roles in postnatal cartilage development and protect against OA-associated cartilage damage. 38 In this study, the genes involved in the FoxO signaling pathway were all downregulated. Modulation of FoxO pathway in synovium may be advantageous for OA amelioration.
Hub genes such as JUN, EGR1, JUND, KLF4, MYC, and CEBPB not only played important roles in the PPI network but also acted as central nodes and transcription factors in the transcriptional regulatory network. For example, JUN and JUND, members of the activator protein-1 family, a group of transcription factors, 39 are involved in cell proliferation, differentiation, apoptosis, and survival via participation in the transcriptional network, and immune responses regulation.40,41 As transcription factors, EGR1, KLF4, MYC, and CEBPB also perform diverse transcriptional functions in many cell physiological functions and cellular processes.42 -45 Overexpressed EGR1 in chondrocytes could accelerate chondrocyte hypertrophy, prevent COl2A1 expression, and promote the release of inflammatory factors, such as MMP9 and MMP13. 46 Notably, endoplasmic reticulum stress can kinetically control CEBPB isoforms in fibroblasts. Furthermore, CEBPB pathways can be involved in antitumor immunity mediated by the myeloid-derived suppressor cells that are inhibited in triple-negative breast cancer. 47 However, the function and mechanism of these transcription factors in OA require further research.
Previous studies have demonstrated strong correlations between the infiltration of immune cells and OA.48,49 The inflammatory membranes of OA synovium are often infiltrated by immune cells. 34 Th17 is an immune cell which is implicated in the pathogenesis of most common autoimmune diseases, including RA. 50 Recent findings have shown that FOSB is an essential regulator that determines the production of transforming growth factor (TGF)-β. 51 TGF-β is a regulator of CD4+ T cell differentiation and determines the balance of Th17 and Treg cells for autoimmunity. 52 KLF6 is a potential regulator of Th17 cells. Iliopoulos et al. 53 suggested that KLF6 is a novel protein that was deregulated in OA. Our results demonstrated that the expression of hub genes was regulated by several transcription factors that are significantly correlated with immune cell infiltration. Further research is required to provide guidance for future biological studies and clinical immunotherapy in OA.
Several genes, such as IL-1β and MMP-13, are coexpressed in cartilage, synovium, and subchondral bone tissues and play important roles in the development of OA.5,54,55 Herein, we used 12 public data sets and experiment validation of mRNA and protein levels to demonstrate that APOD is a hub gene coexpressed and downregulated in the diseased condition as compared to normal tissue. Tew et al. 56 demonstrated that APOD expression was downregulated not only in patients with OA but also in the highly loaded and more physically damaged cartilage. Furthermore, APOD was downregulated in OA cartilage and synovium, as previously demonstrated.57 -61 In animal experiments, APOD was also correlated with OA in synovial fluid samples and chondrocytes.62,63 APOD is a secreted glycosylated protein and an atypical lipoprotein, which can bind to several small molecules, such as arachidonic acid, steroids, and cholesterol. 64 APOD is neuro- and cardio-protective and has important functions such as oxidation resistance, anti-inflammation, and stress resistance. 65 Furthermore, in aged mice, APOD deficiency can cause high bone turnover, low bone mass, and impair osteoblastic function. 66 Although in the small samples included in this study, the lower APOD levels correlated with OA, it remains to be confirmed in a large sample size. Also, further investigations into the potential role and underlying mechanism of APOD in OA development remain warranted.
Conclusion
The most significant module associated with OA trait was identified based on the WGCNA method for synovium tissue transcription analysis, and APOD was validated as a hub gene coexpressed and downregulated in multiple tissues. Furthermore, transcriptional regulatory-immune network was constructed, which may help to improve immune therapy for OA.
Supplemental Material
sj-csv-1-car-10.1177_19476035211053824 – Supplemental material for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis
Supplemental material, sj-csv-1-car-10.1177_19476035211053824 for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis by Yong Qin, Jia Li, Yonggang Zhou, Chengliang Yin, Yi Li, Ming Chen, Yinqiao Du, Tiejian Li and Jinglong Yan in CARTILAGE
Supplemental Material
sj-csv-2-car-10.1177_19476035211053824 – Supplemental material for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis
Supplemental material, sj-csv-2-car-10.1177_19476035211053824 for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis by Yong Qin, Jia Li, Yonggang Zhou, Chengliang Yin, Yi Li, Ming Chen, Yinqiao Du, Tiejian Li and Jinglong Yan in CARTILAGE
Supplemental Material
sj-cys-1-car-10.1177_19476035211053824 – Supplemental material for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis
Supplemental material, sj-cys-1-car-10.1177_19476035211053824 for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis by Yong Qin, Jia Li, Yonggang Zhou, Chengliang Yin, Yi Li, Ming Chen, Yinqiao Du, Tiejian Li and Jinglong Yan in CARTILAGE
Supplemental Material
sj-cys1-car-10.1177_19476035211053824 – Supplemental material for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis
Supplemental material, sj-cys1-car-10.1177_19476035211053824 for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis by Yong Qin, Jia Li, Yonggang Zhou, Chengliang Yin, Yi Li, Ming Chen, Yinqiao Du, Tiejian Li and Jinglong Yan in CARTILAGE
Supplemental Material
sj-docx-1-car-10.1177_19476035211053824 – Supplemental material for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis
Supplemental material, sj-docx-1-car-10.1177_19476035211053824 for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis by Yong Qin, Jia Li, Yonggang Zhou, Chengliang Yin, Yi Li, Ming Chen, Yinqiao Du, Tiejian Li and Jinglong Yan in CARTILAGE
Supplemental Material
sj-docx-2-car-10.1177_19476035211053824 – Supplemental material for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis
Supplemental material, sj-docx-2-car-10.1177_19476035211053824 for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis by Yong Qin, Jia Li, Yonggang Zhou, Chengliang Yin, Yi Li, Ming Chen, Yinqiao Du, Tiejian Li and Jinglong Yan in CARTILAGE
Supplemental Material
sj-docx-3-car-10.1177_19476035211053824 – Supplemental material for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis
Supplemental material, sj-docx-3-car-10.1177_19476035211053824 for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis by Yong Qin, Jia Li, Yonggang Zhou, Chengliang Yin, Yi Li, Ming Chen, Yinqiao Du, Tiejian Li and Jinglong Yan in CARTILAGE
Supplemental Material
sj-pdf-1-car-10.1177_19476035211053824 – Supplemental material for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis
Supplemental material, sj-pdf-1-car-10.1177_19476035211053824 for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis by Yong Qin, Jia Li, Yonggang Zhou, Chengliang Yin, Yi Li, Ming Chen, Yinqiao Du, Tiejian Li and Jinglong Yan in CARTILAGE
Supplemental Material
sj-xlsx-1-car-10.1177_19476035211053824 – Supplemental material for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis
Supplemental material, sj-xlsx-1-car-10.1177_19476035211053824 for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis by Yong Qin, Jia Li, Yonggang Zhou, Chengliang Yin, Yi Li, Ming Chen, Yinqiao Du, Tiejian Li and Jinglong Yan in CARTILAGE
Supplemental Material
sj-xlsx-2-car-10.1177_19476035211053824 – Supplemental material for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis
Supplemental material, sj-xlsx-2-car-10.1177_19476035211053824 for Apolipoprotein D as a Potential Biomarker and Construction of a Transcriptional Regulatory-Immune Network Associated with Osteoarthritis by Weighted Gene Coexpression Network Analysis by Yong Qin, Jia Li, Yonggang Zhou, Chengliang Yin, Yi Li, Ming Chen, Yinqiao Du, Tiejian Li and Jinglong Yan in CARTILAGE
Footnotes
Author Contributions
Jinglong Yan, Yong Qin, and Yonggang Zhou conceived and designed the research study; Chengliang Yin and Yong Qin performed data analysis; Jia Li, Yi Li, Ming Chen, Yinqiao Du, and Tiejian Li collected patient samples and performed the experimental validation; Yong Qin drafted the original article; Jinglong Yan and Yonggang Zhou revised the article. All authors read and approved the final article to be published.
Acknowledgments and Funding
We thanked Professor Desi Shang for providing metagene that represented 28 immune cell types. The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (grant number:82072472) and Medical Big Data and AI R&D Project of Chinese PLA General Hospital (2019MBD-001).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
This study was approved by the Ethics Committee of the Second Affiliated Hospital of Harbin Medical University (ethical approval number: ky2020-078). Written informed consent was obtained from each tissue donor included in this study.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.