
Abstract
Objective
Hypertrophic cartilage formation is a major setback in mesenchymal stem cells (MSCs)–mediated cartilage repair, and overcoming it requires optimization of differentiation. Here, we tested the miR-140 activated collagen hydrogel for the chondrogenic differentiation of MSCs and to produce hyaline cartilage.
Methods
Bone marrow MSCs isolated from 3 patients were pretreated with miR-140 and then chondrogenic differentiated. The 3-dimensional (3D) transfection potential of 5 different transfection reagents (Polyethylenimine, Lipofectamine, TransIT-X2, Amide:Cholesterol-based liposomes [AmC] and AmC pegylated with Tocofersolan [AmCTOC]) was compared and the reagent that showed higher green fluorescent protein (GFP) expression was selected. Finally, the collagen hydrogel was activated using miR-140-transfection complex and sustained delivered to MSCs during chondrogenic differentiation. After differentiation, the outcome was assessed by reverse transcription–polymerase chain reaction (RT-PCR), histology, immunohistochemistry, and compared with scrambled miRNA treated control.
Results
Pretreatment of MSCs with miR-140 significantly increased the expression of cartilage-specific genes (COL2A1, SOX9, and ACAN) with reduced hypertrophic chondrocyte (COL10A1) marker expression and better safranin-O staining than the control. The AmCTOC liposome showed a significant increase in 3D transfection of GFP expressing plasmid than the others. Furthermore, the knockdown of GAPDH using siRNA in HEK cells and expression of GFP mRNA in human bone marrow MSCs confirmed the 3D-transfection efficiency of AmCTOC. The sustained delivery of miR-140 using activated matrix formed a hyaline cartilage-like tissue with minimal COL10A1 expression in RT-PCR and immunohistochemistry.
Conclusion
Our results demonstrated the therapeutic potential of miR-140-activated hydrogel for MSCs-based cartilage tissue engineering, which could also be used for endogenous stem cells–mediated cartilage repair.
Keywords
Introduction
Complete repair of articular cartilage (AC) defect is an unresolved clinical problem considering its poor self-healing ability and paucity of healthy cartilage for the treatment. Mesenchymal stem cells (MSCs) and their multilineage potential offer an excellent substitute, and they can be easily sourced from many tissues. The preclinical and clinical studies have illustrated that implantation of MSCs has the potential to resurface the cartilage defect, but their success is limited by their propensity to become hypertrophic chondrocytes.1,2 The hypertrophic chondrocytes produce transient cartilage, which typically undergoes endochondral ossification and forms bone instead of cartilage. 3 To attain the hyaline cartilage and avoid hypertrophy, it necessitates a protocol for controlled differentiation of MSCs to chondrocytes and retains the cells in stable cartilage phenotype.
MicroRNAs (miRNAs) are pivotal in regulating the chondrogenesis of MSCs. They are small noncoding RNAs that post-transcriptionally regulate the gene expression by inhibiting the translation or the degradation of complementary mRNA. Among the different miRNAs, miR-140 is most often studied because it is expressed explicitly in the AC and enables hyaline phenotype. The overexpression of miR-140 using lentiviral vectors can induce the chondrogenic differentiation of stem cells. 4 Geng et al. 5 showed the intra-articular injection of miR-140 overexpressing umbilical cord MSCs enhanced the hyaline cartilage regeneration in the surgically induced osteoarthritis (OA) rat model. Viral vectors are commonly used to demonstrate the proof of concept. However, the tumorigenicity and their tendency to elicit an immune response are significant concerns preventing their clinical translation. Alternatives such as an exosome-mediated delivery of miR-140 have shown promising chondrogenesis of MSCs and reduced OA manifestations.6,7 The exosome-mediated or nonviral vector delivery is relatively safe, but it requires multiple injections as their effect is transient.
The miRNA-activated matrix (3-dimensional [3D] system) offers a safe and sustained delivery solution for miRNA to differentiate the stem cells and facilitate tissue regeneration. The miRNA-activated matrix comprises the therapeutic miRNA complexed with the transfection reagent and encapsulated/loaded in the scaffold. The construct slowly releases the complex, which transfects and modifies the adjacent cells favorably. Besides, the scaffold material provides structural architecture and a niche for regenerating tissue. The miRNA-activated matrices have been shown to induce osteogenic differentiation in MSCs and potentiate the endogenous stem cells to repair bone defects.8,9 The activated matrices have been broadly tested for tissue engineering applications such as bone and blood vessels.10,11 Nevertheless, 2 prior reports from Lolli et al.12,13 have explored the miR-221 activated matrix for AC engineering using a lipofectamine-based transfection reagent, which is shown to cause cytotoxicity.14,15 Furthermore, MSCs exhibits poor transfection potential, and the process is further limited in 3D condition. 16 Hence there is scope for further exploration of safe and effective activated matrices to create hyaline cartilage tissue.
The study aimed to generate hyaline cartilage from human bone marrow MSCs (BM-MSCs) using sustained delivery of miR-140 from an activated matrix. Toward developing a nucleic acid–activated matrix, we used 2-hydroxy-N-methyl-N,N-bis(2-tetradecanamidoethyl)ethanaminium chloride (amide linker–based cationic lipid) to develop the liposomal formulations. Our prior findings demonstrated that liposomes of amide linker–based were efficient in delivering nucleic acids, including pDNA, miRNA, and siRNA in vitro and in vivo.17-19 Taking cues from these findings, we developed liposomes with amide linker–based cationic lipid, cholesterol as colipids and pegylated with Tocofersalon to improve its transfection potential. Subsequently, we explored the usefulness of our novel liposomal formulation for 3D transfection and compared the transfection efficiencies with commercially available transfection agents.
Materials and Methods
Isolation and Characterization of BM-MSCs from Trabecular Bone
This study received the institutional review board approval and was conducted as per the recommendation of the Declaration of Helsinki. The patient samples were collected after obtaining informed written consent. Trabecular bone was obtained from 3 patients, mean age 4 ± 1.73 years, undergoing closing wedge corrective osteotomy surgeries. Human BM-MSCs were isolated from the trabecular bone as described previously. 20 The isolated cells were cultured with MSC culture medium (α-minimum essential medium [αMEM] supplemented with 10% fetal bovine serum [FBS], 5 ng/mL fibroblast growth factor-2 [FGF-2]; (PeproTech, Rocky Hill, NJ), 50 µg/mL gentamicin, and 2µg/ml amphotericin b). Cells were expanded up to the third passage and used for characterization and further studies. The phenotype of cells was confirmed as per the criteria set forth by the International Society for Cellular Therapy (multilineage differentiation; and CD surface markers expression for plastic adherent fibroblast-like cells). This study’s reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified. Experiments on BM-MSCs were performed with 3 biological samples and each with 2 technical replicates.
Two-Dimensional Transfection of MSCs with miR-140 and Chondrogenic Differentiation in Pellet Culture
First, the third passaged MSCs were cultured in a 24-well plate at a seeding density of 6 × 104 cells/well. Twenty-four hours later, cells were transfected with 20 nM miRCURY LNA miR-140 Mimic (Qiagen, Hilden, Germany) using TransIT-X2 Dynamic Delivery System (Mirus Bio LLC, Madison, WI, USA) as per the manufacturer’s protocol. After 24 hours of incubation, the transfection medium was replaced with the MSC culture medium without FGF-2 and cultured for 48 hours. Cells were transfected a second time using the above protocol.
Forty-eight hours after transfection, cells were trypsinized and seeded in a 96-well plate at a high density (1 × 105 cells/well) to form a pellet. The pellet was cultured with a chondrogenic differentiation medium (Dulbecco’s modified Eagle medium [DMEM]–high glucose [HG]; 4.5 g/mL supplemented with 10 ng/mL human transforming growth factor-β3, 1 mM sodium pyruvate, 40 μg/mL ascorbate-2-phosphate, 10−7 M dexamethasone, 1% ITS+1, and 40 µg/mL
Preparation of Liposomal Formulations
Amide lipid was synthesized and characterized as per the previously published protocol. 17 The cationic liposomes, Amide:Cholesterol (AmC), and Amide:Cholesterol:Tocofersolan (AmCTOC), were prepared using the thin-film hydration method. Protocols for preparation of liposomes and characterization are given in the Supplemental Material.
Selection of a Transfecting Agent
The 3D transfection ability of the 2 in-house prepared reagents (AmC and AmCTOC) were tested and compared with 3 commercially available reagents, namely branched polyethylenimine (BPEI), TransIT-X2 Dynamic Delivery System, and Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). A 9.75-kb plasmid pNL-EGFP/CMV/WPREdU3 (Addgene plasmid 17579) containing green fluorescent protein (GFP) gene was transfected into the human embryonic kidney 293 cells (HEK293) in a collagen hydrogel (Invitrogen, Carlsbad, CA, USA). The best performing transfection reagent was chosen for further studies. All 5 transfection complexes were prepared in a 2% sucrose solution. At first, 5 µg of plasmid was diluted in 250 µL sucrose solution and incubated for 5 minutes at room temperature. The transfection reagents (7.5 µL BPEI [1 mg/mL]/12.5 µL TransIT-X2/10 µL Lipofectamine 3000/15 µL AmC or AmCTOC) were added to the respective tube, and the tubes were immediately vortexed and incubated at 37 °C for 20 minutes to form the transfection complex. The plasmid-transfection solutions were maintained at −80 °C for 2 hours before overnight freeze-drying (Martin Christ alpha 1-2 LD plus, Germany). The lyophilized complexes were suspended in nuclease-free water.
Meanwhile, HEK cells at 90% confluence were trypsinized, and the cell pellet was resuspended at a cell density of 3 × 105 cells/10 µL (medium). The lyophilized complex and HEK cells were mixed with collagen (final concentration of 2% [w/v]) hydrogel as per the manufacturer’s protocol. After polymerization, the hydrogels were transferred to an Ultra-Low Attachment culture plate (Corning, NY, USA) and cultured with DMEM-HG medium supplemented with 10% FBS. On the 7th, 14th, and 21st day of culture, the hydrogels were observed under a confocal microscope (Olympus FV1000). The transfection reagent that has higher 3D transfection potential and produces a large number of GFP expressing cells was selected for further experiment. Three independent screenings (each composed of duplicates) were performed to confirm the 3D transfection potential of the selected reagent.
The enhanced green fluorescent protein (eGFP) mRNA (990bp) (Trilink Biotechnologies, San Diego, CA) was used to confirm the 3D transfection of human BM-MSCs. The transfection complex containing 3µg of eGFP mRNA and 9 µL of transfection reagent was prepared and lyophilized, as mentioned above. The human MSCs (3 × 105 cells) and the lyophilized complex were encapsulated in collagen hydrogel and cultured with the MSC culture medium without antibiotics. After 7 days of culture, the construct was stained with ethidium homodimer (LIVE/DEAD Viability/Cytotoxicity Kit, Invitrogen, Carlsbad, CA, USA) and 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen, Carlsbad, CA, USA) and imaged using a confocal microscope (Olympus FV1000).
Release Kinetics of Collagen Hydrogel
The release kinetics of collagen hydrogel was studied using Alexa Fluor 546 labeled All-Stars Negative Control siRNA (Qiagen, Hilden, Germany) and the detailed protocol is given in the Supplemental Material.
Bioactivity of siRNA on Hydrogel
Bioactivity of transfection complex on hydrogel was determined using siRNA targeting GAPDH (siGAPDH, RIBOBIO, Guangzhou, China). The transfection complex was prepared using 3 µg of siGAPDH and 9 µL of AmCTOC and then encapsulated with HEK (3 × 105 cells) in collagen hydrogel. The fabricated hydrogel was transferred to an Ultra-Low Attachment culture plate and cultured with antibiotic-free DMEM-HG medium supplemented with 10% FBS. The samples were collected on different days of culture to assess the knockdown of GAPDH expression in HEK cells using gene expression analysis. The outcome was compared with the control hydrogel loaded with nontargeting control siRNA (All-Stars Negative Control siRNA; Qiagen, Hilden Germany). Bioactivity experiment was performed with 3 technical triplicates.
Sustained Delivery of miR-140 during Chondrogenic Differentiation
The preparation of the miR-140-AmCTOC transfection complex and loading of these complexes with human MSC (3 × 105) in collagen hydrogel was performed as mentioned above. The construct was cultured in the chondrogenic differentiation medium for 3 weeks. The quality of the resulting tissue-engineered cartilage construct was assessed by histology, immunohistochemistry, and gene expression analysis. The differentiated cells on hydrogel with no target scrambled control served as a comparison.
Histology and Immunohistochemistry
The cartilage pellets were fixed in HistoChoice tissue fixative solution for 12 hours at room temperature and embedded in paraffin wax by following the standard histological protocol. Five-micrometer sections were prepared from the tissue block and stained with safranin-O dye to visualize glycosaminoglycan (GAG) dissemination. For immunohistochemistry, the tissue sections were stained using mouse anti-collagen type 2 (Developmental Studies Hybridoma Bank, II-II6B3, dilution 1:5) or rabbit anti-collagen type 10 antibody (Merck, 234196, dilution 1:250). The EnVision FLEX detection system (DAKO, Glostrup, Denmark) was used to develop the color and then observed under a light microscope (Leica DM 2000). The percentage of area stained with anti-collagen type 2 and anti-collagen type 10 antibodies were semiquantitated using Image J software version 1.53e (National Institutes of Health, Bethesda, MD, USA).
Reverse Transcription–Polymerase Chain Reaction Analysis
The total RNA was isolated from the sample using TRI Reagent as per the manufacturer’s protocol and then converted to cDNA using a SuperScript VILO cDNA Synthesis Kit (Invitrogen, Carlsbad, CA, USA). The expression of chondrocyte-specific markers (COL2A1, SOX9, and ACAN) and hypertrophic marker (COL10A1) were quantitated using Taqman gene expression assays (Invitrogen, Carlsbad, CA, USA). The beta-actin was used as a reference gene, and the relative gene expression was calculated by the ΔΔCt method and represented in fold change.
Statistics
Unpaired Student t test was performed to test the significance between the miR-140 mimics treated and the control sample. One-way analysis of variance with Tukey multiple comparison post hoc test was used to compare the transfection efficiency of different transfection reagents. Data were represented as mean ± SD. A calculated P value of less than 0.05 was considered significant.
Results
Isolation and Characterization of BM-MSCs
BM-MSCs were isolated from 3 trabecular bone samples and expanded up to 3 passages in monolayer culture. Subsequent flow cytometry analysis showed that the third passaged cells were stained >99% positive for MSC markers (CD 73, CD 90, CD 105), and <3% cells for hematopoietic markers (CD 14, CD 34, CD 45) (see Supplemental Figure S1).
Multilineage Differentiation
Functional characteristics of isolated cells were confirmed by multilineage differentiation. Three weeks after differentiation, chondrogenesis was demonstrated by GAG deposition in the cartilage pellet using safranin-O staining. Alizarin red staining was performed, and it illustrated calcium deposition in the culture, confirming osteogenic differentiation. Oil O red staining of lipid droplets in the culture showed the adipogenic differentiation of MSCs (see Supplemental Figure S2). Together, these findings confirmed that the isolated cells exhibited multipotency.
miR-140 and Chondrogenic Differentiation
BM-MSCs were transfected with miR-140 mimics and then subjected to chondrogenic differentiation. Gene expression analysis at three weeks of differentiation showed a significant increase in COL2A1 (1.78-fold), SOX9 (1.43-fold), and ACAN (3.09-fold) expression in the miR-140-treated samples compared with the control ( Fig. 1A ). Besides, the expression of COL10A1 (0.74-fold) was lesser in miR-140-treated samples than in control.
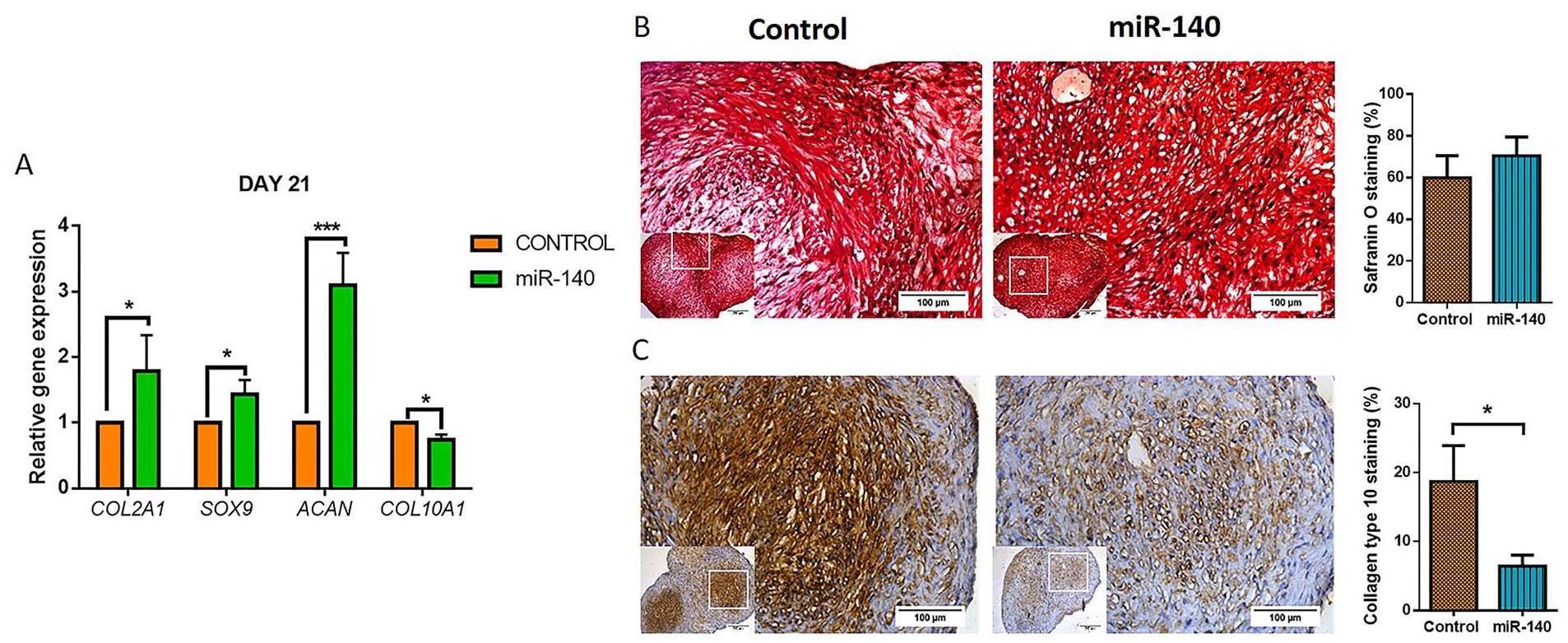
(
Safranin-O staining was performed to assess the cartilage derived from BM-MSCs qualitatively. The control group had lesser safranin-O staining (P = ns) than the miR-140-treated sample ( Fig. 1B ). Further analysis of collagen type 10 immunostaining showed that the intensity of staining was higher in the control group and corresponded to the areas showing reduced staining of safranin O. In contrast, the miR-140-treated samples showed significantly (P < 0.05) lesser collagen type 10 staining than the control, suggesting lesser hypertrophic chondrocytes differentiation ( Fig. 1C ). Collectively, the gene expression, histology, and immunostaining analyses confirmed that the miR-140 promotes hyaline-like cartilage formation and inhibits the hypertrophic differentiation of MSCs.
Characterization of Liposomal Formulations
Liposomal formulations were prepared using a previously reported transfection efficient cationic amphiphile, amide linker lipid with neutral lipid, cholesterol (AmC). We also prepared AmC liposome pegylated with various Tocofersolan concentrations (0.25 to 1 mM) (AmTOC 1-4). The dynamic light scattering analysis showed that the hydrodynamic diameters of AmC liposomes were around 230 nm, and increasing the concentration of the Tocofersolan increased the sizes of the liposomes from 290 to 550 nm (see Supplemental Figure S3). The AmC with 0.25 mM of Tocofersolan (AmCTOC-1) showed a slight increase in the size from 230 to 290 nm. Further increasing the Tocofersolan from 0.5 to 1mM concentrations increased the sizes from 480 to 550 nm. The surface potential of AmC was +21 meV, whereas the addition of Tocofersolan diminished the surface charge. Tocofersolan at 0.25 mM did not affect surface charges significantly (+17 meV) but in higher concentration, the surface charge reduced to +12 meV at 0.5 mM, +10 meV at 0.75 mM, and +6 meV at 1 mM. As expected, pegylation increased the size of the liposomes due to the hydration effect and decreased the surface charge due to the shielding of polyethylene glycol molecules. Considering the optimal physicochemical characteristics of the liposomes, we have chosen AmC and AmCTOC-1 (hereafter referred to as AmCTOC) for our further studies.
The transmission electron microscope analysis illustrated both AmC and AmCTOC liposomes were spherical vesicles with an average diameter of 128.7 ± 24.9 nm and 151.3 ± 51.2 nm, respectively ( Fig. 2 ). When they were complexed with GFP plasmid (9.75 kb), sizes of both AmC and AmCTOC lipoplexes decreased to a diameter of 72.2 ± 15.3 nm and 89.8 ± 17.1 nm, respectively. The AmC-lipoplex formed aggregates, whereas AmCTOC-lipoplex remained discrete, evenly distributed, and retained their shape ( Fig. 2 ). AmcTOC formed a thicker film on the copper grid as compared with AmC, causing higher background noise.
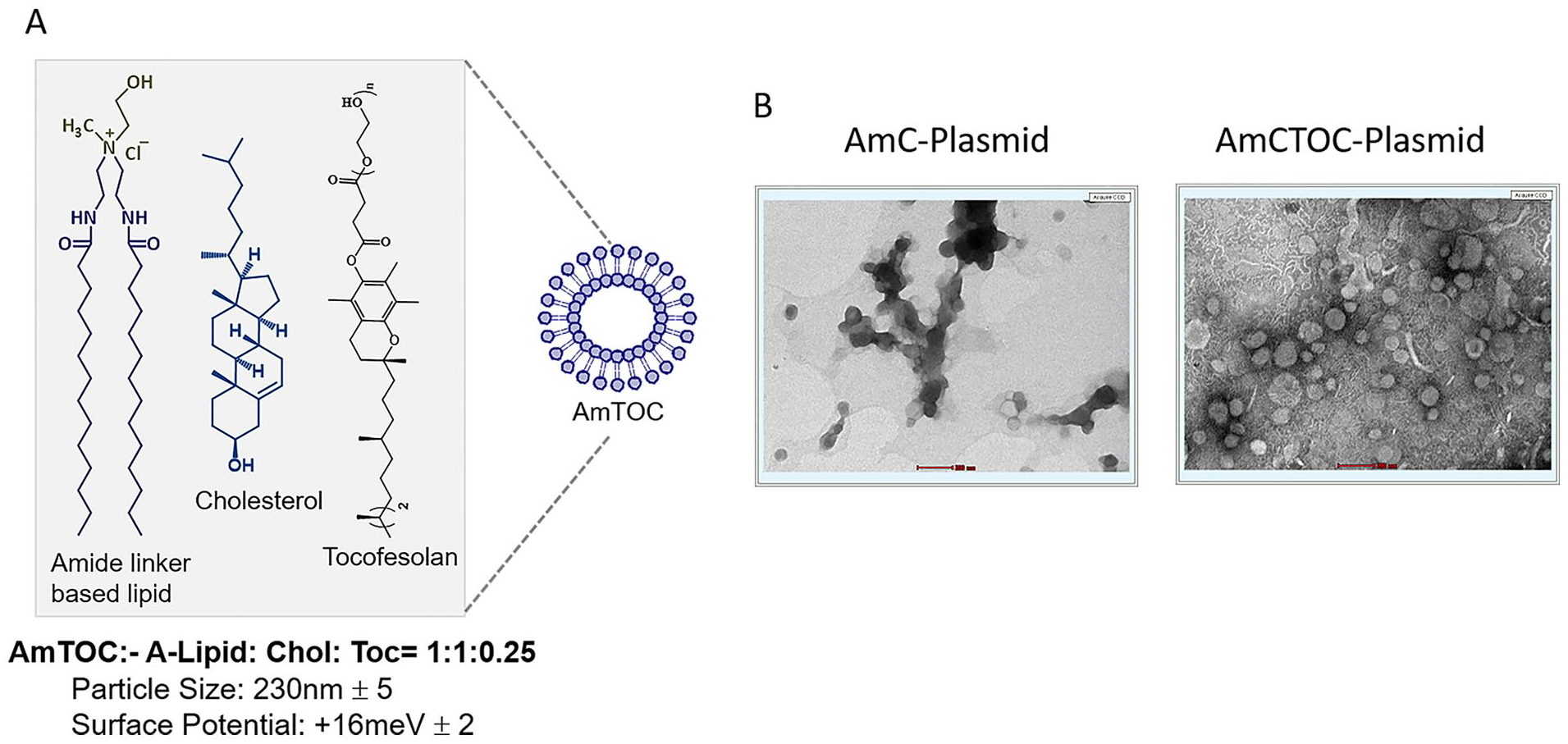
(
Selection of Transfection Reagent
Five cationic transfection reagents were screened in this study to find an ideal reagent for transfecting cells in the 3D condition. Confocal analysis on the day 7 sample showed that the AmCTOC-lipoplex exhibited significantly (P < 0.001) higher transfection efficiencies. A larger number of GFP-expressing cells were observed in AmCTOC transfected group compared with the other groups transfected with either Lifectamine/BPEI/TransIT or AmC transfection reagent ( Fig. 3 ). On 14th and 21st day, both AmCTOC and Lipofectamine 3000 showed significantly higher transfection than the other 2 groups; compared with Lipofectamine 3000, the AmCTOC showed higher transfection (P = ns) efficiency and therefore selected for further testing. The number of GFP expressing cells decreased with the culture duration in all 5 groups.
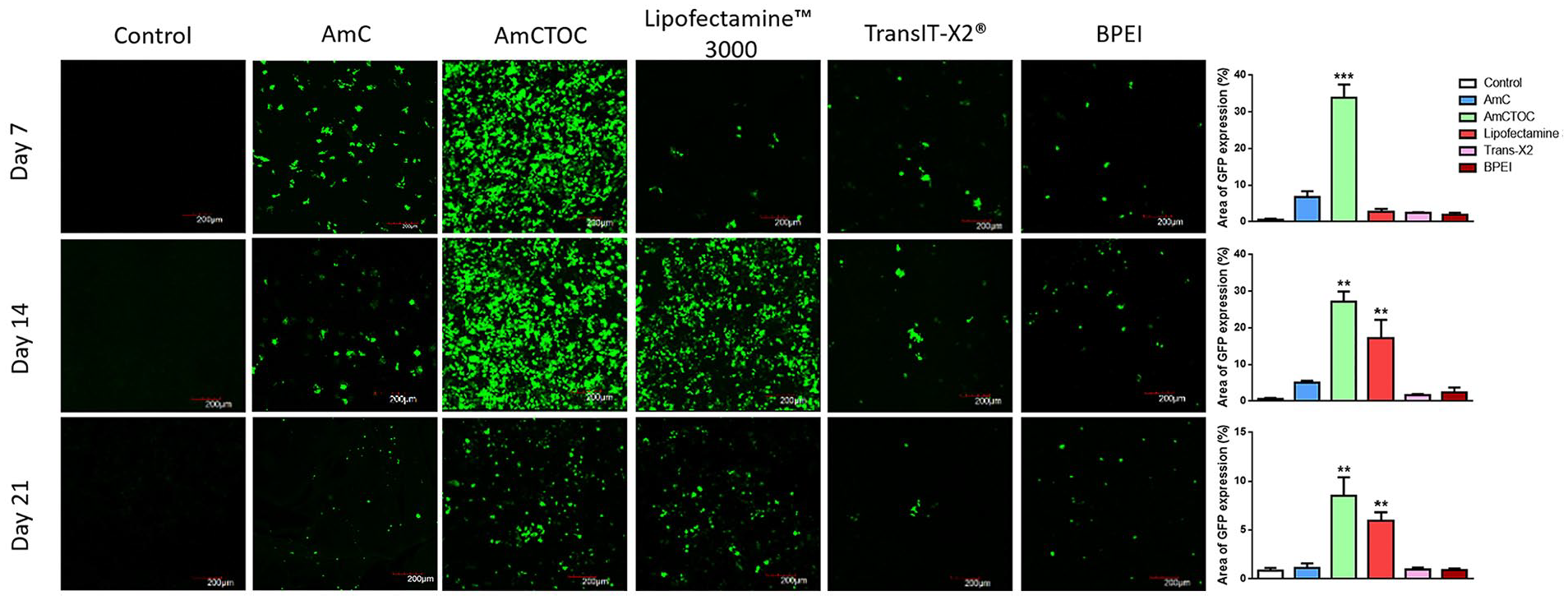
Confocal microscopic images show the green fluorescent protein (GFP) expression in the HEK cells on the 7th, 14th, and 21st day (n = 3 experiments with duplicates in each). The green color represents the GFP expressing cells. Quantification using Image J software (bar diagram) confirmed that AmCTOC promoted higher 3D transfection and exhibited a large number of GFP expressing cells than the other groups. **P < 0.01, ***P < 0.001.
Release Kinetics of Collagen Hydrogel
The release profile of nucleic acids in collagen hydrogel was evaluated using a fluorescently labeled siRNA. The sustained release of the AmCTOC-siRNA complex from hydrogel was detected for up to 3 weeks in phosphate buffered saline at 37 °C ( Fig. 4A ). The release profile began with an initial burst release of 49.02% siRNA complex by the third day and reached 80% cumulative release within 1 week of incubation. During the second week, 359.6 ± 24.16 ng on day 9, 156.56 ± 65.15 on day 12, and 95.49 ± 7.63 of siRNA complex on day 15 were released ( Fig. 4B ). At the end of the second week, 92% of the payload was released from the hydrogel, and then the release kinetics reached the lag phase during the third week of incubation. This outcome demonstrated that the decreased GFP-expressing cells on day 14 and day 21 of culture was due to the lesser amount of transfection complex in the hydrogel, and most of it was released within the first week of culture.
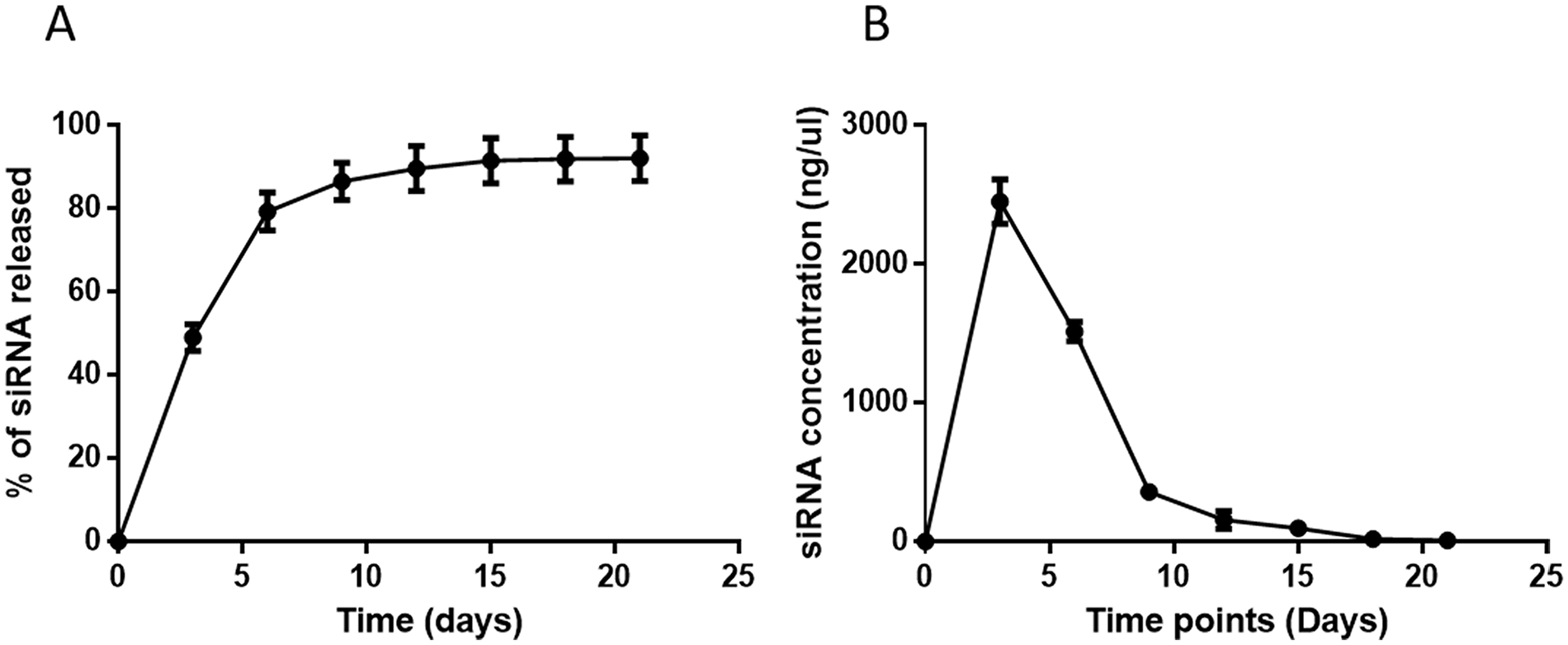
(
Bioactivity of Transfection Complex in Hydrogel
The function and stability of the transfection complex in the hydrogel were confirmed using siGAPDH. Gene expression analysis showed GAPDH expression in HEK cells was significantly (P < 0.001) reduced by 2.32-fold on day 3 and 1.74-fold on day 7 in the siGAPDH-treated group compared with the control. Further analysis on day 14 samples showed GAPDH expression was 1.35-fold lesser in the siGAPDH group than in the control ( Fig. 5A ). These results confirmed that the bioactivity of the transfected complex remained functional during the release period.
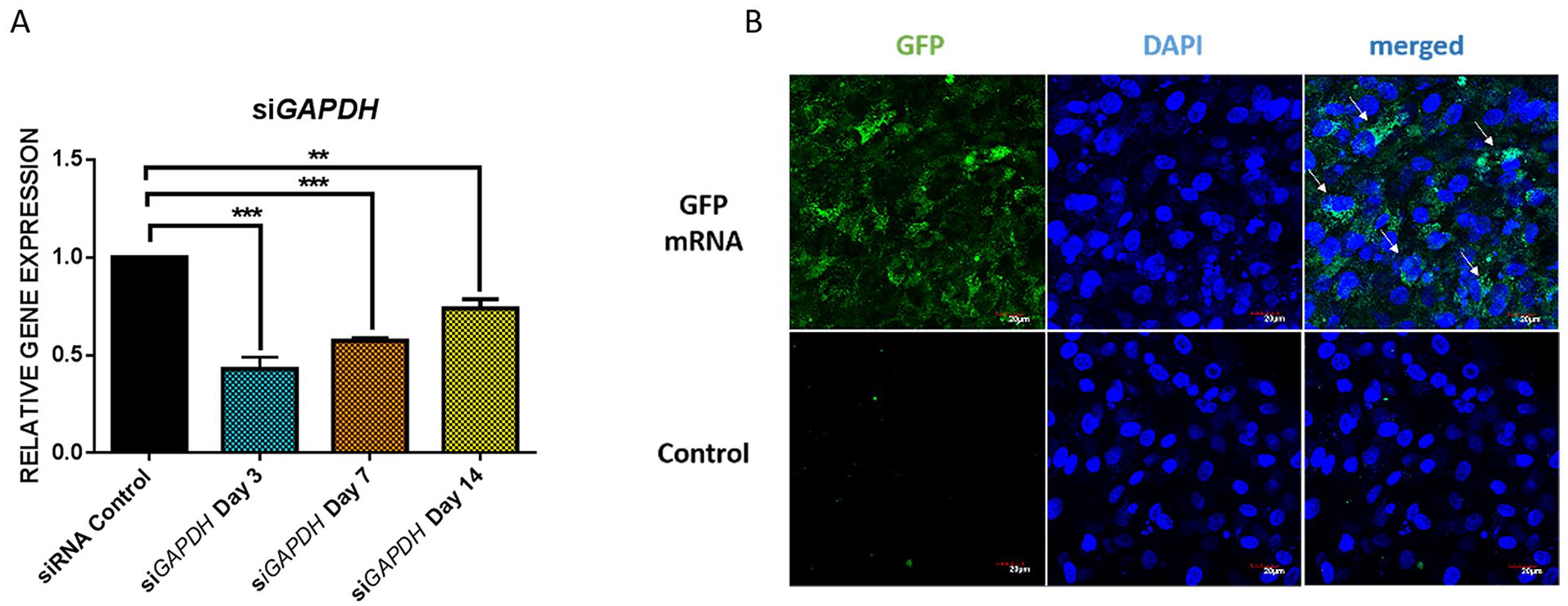
(
Subsequently, the 3D transfection efficiency of the AmCTOC reagent was reconfirmed using mRNA expressing GFP transcript on human BM-MSCs. After seven days of transfection, confocal microscopy showed 87.1% ± 5.6% of cells expressed GFP, confirming mRNA uptake by human BM-MSCs ( Fig. 5B and Supplemental Figure S4). Cytotoxicity analysis confirmed that the 3D transfection using AMCTOC was not toxic, and there were no dead cells appeared in the ethidium homodimer (EthD) nuclear staining (see Supplemental Figure S4).
Sustained Delivery of Selected miRNA during Chondrogenic Differentiation
After establishing the 3D transfection protocol, the miR-140 was complexed with AmCTOC and then loaded on hydrogels with human MSCs. After chondrogenic differentiation, gene expression analysis showed that the miR-140-treated group had significantly higher COL2A1, SOX9, and ACAN expression on day 14 and day 21 compared to the scrambled delivered control. The expression of hypertrophic marker COL10A1 was significantly reduced in the miR-140-treated group compared with the control ( Fig. 6 ).
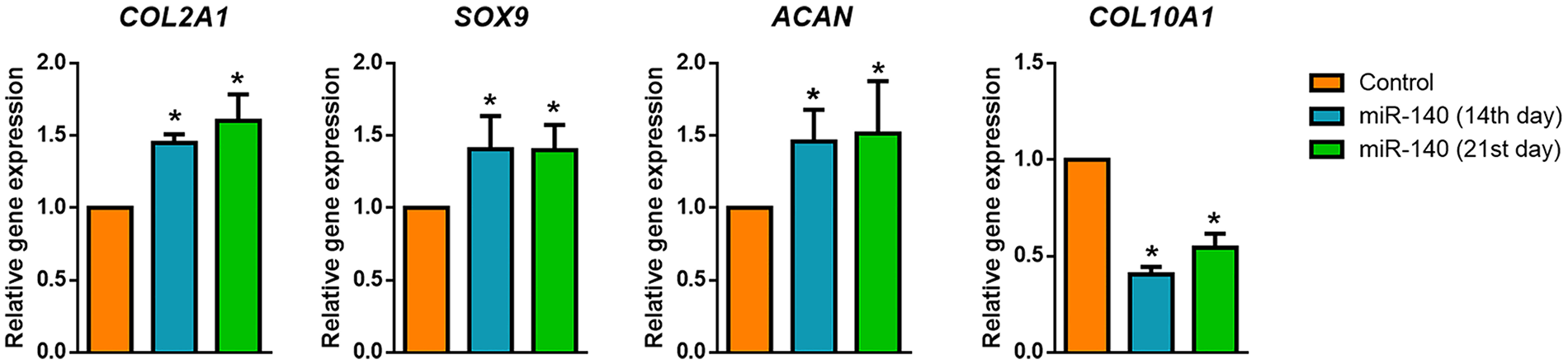
Cartilage-specific genes (COL2A1, SOX9, and ACAN) and chondrocyte hypertrophic marker (COL10A1) expression in the miR-140 compared with control scrambled miRNA delivered samples (n = 3 independent experiment and each performed in duplicate). The sustained delivery of miR-140 induced higher chondrogenic differentiation with less hypertrophic marker expression than the control on both the 14th and 21st days of differentiation. *P < 0.05.
Histology and Immunohistochemistry
After 21 days of chondrogenic differentiation, histological analysis for GAG staining showed both the miR-140 and scrambled miRNA treated samples stained equally well with safranin-O dye ( Fig. 7A ). Qualitatively, there was no difference between the 2 groups except in a few places, where the scrambled control showed lesser staining.
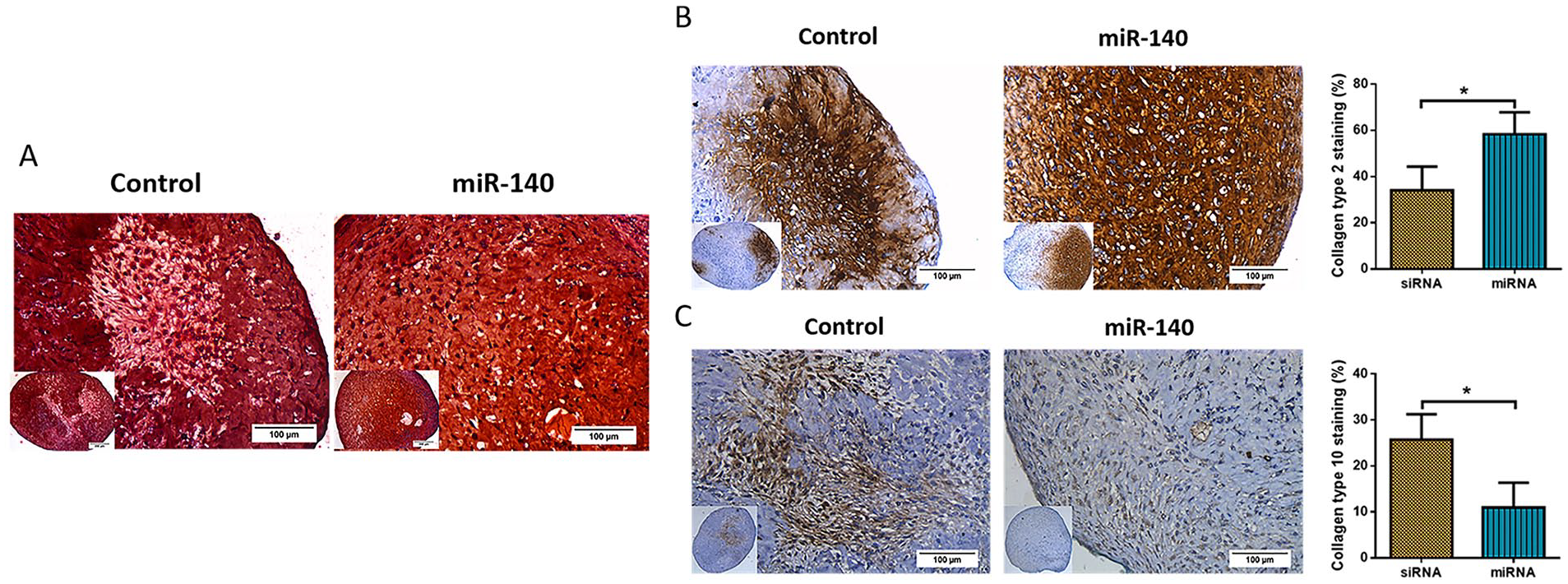
Light microscopic images of tissue-engineered cartilage from mesenchymal stem cells (MSCs) using miR-140-activated collagen matrix. (
Collagen type 2 and collagen type 10 immunostaining were performed to evaluate the chondrogenic and hypertrophic chondrocytes differentiation. The outcome showed that the collagen type 2 staining was significantly higher in the miR-140 treated sample than the control ( Fig. 7B ). Furthermore, the miR-140 treated sample showed a lesser collagen type 10 staining than the control ( Fig. 7C ).
Thus, the gene expression and immunostaining outcomes quantitatively confirmed that the sustained delivery of miR-140-AmCTOC complex through the collagen hydrogel significantly enhanced the hyaline cartilage formation from MSCs and inhibited hypertrophic differentiation when compared with the scrambled control.
Discussion
Molecular mechanisms regulating the chondrogenesis of MSCs are the key to understand hyaline cartilage formation, and they may aid in developing an ideal therapeutic strategy. The miRNAs are one such factor that is critically involved in cartilage development and conserve the stable hyaline phenotype. Most reported studies test their function and downstream targets, but rarely explore their effect on tissue regeneration. Furthermore, a safe and efficient delivery system is essential to use these biomolecules effectively in clinical translation. We have attempted to harness the therapeutic potential of miR-140 for AC tissue regeneration and demonstrated a hyaline cartilage tissue formation using miRNA-activated matrix.
We first confirmed the anti-hypertrophic function of miR-140 in monolayer culture, and this was achieved using a well-characterized human BM-MSCs and commercial transfection agent. The hypertrophic chondrocytes were generated using TGF-β3, an often-used growth factor for chondrogenic differentiation. As predicted, the control cartilage pellets showed a higher level of collagen type 10 expression and loss of GAG staining, and these observations are in line with the previous report. 21 Our experiment showed that the pretreatment of MSCs with miR-140 mimics significantly dampened the expression of hypertrophic marker and simultaneously induced a stable cartilage phenotype with higher COL2A1, SOX9, and ACAN expression. Previous studies demonstrated that the miR-140 downregulates the chondrocyte hypertrophy promoting genes (HDAC4 and SMAD1), matrix-degrading enzymes (MMP13 and ADAMTS-5), and senescent genes (p16 and p21).22,23 At the same time, it also represses the RALA, a negative regulator of SOX9, resulting in increased chondrogenesis and stabilization of the hyaline phenotype. 24 Thus, the multifaceted role of miR-140 makes it an ideal therapeutic candidate for cartilage tissue regeneration.
Then we attempted the 3D transfection of miR-140 using activated collagen hydrogel. However, the preliminary screening using commercial transfection agents showed a poor transfection on the hydrogel. To overcome this, we screened 6 different liposomes from our lab (data not shown) and identified that the AmC liposome transfected the human MSCs at high efficiency in monolayer. However, on 3D, it failed to deliver the cargo similar to the commercial reagents. Generally, the preparation of the activated matrix requires the lyophilization of the transfection complex, which is a rate-limiting step and influences the bioactivity of liposomes. 25 Previous studies have also demonstrated that lyophilization promotes vesicular fusion of liposomes that limit its function. 26 We then added carbohydrates as a lyoprotectant to shield the liposomes from aggregation and to preserve their integrity. 27 But, supplementing with sucrose during freeze-drying did not improve the liposome efficiency. Eventually, we pegylated the liposomes with Tocofersolan; a water-soluble, polyethylene glycol derivative of α-tocopherol, which successfully prevented the liposome aggregation and maintained their homogeneity. Further testing on 3D condition showed its transfection superiority over AmC and commercial reagents. These observations were further strengthened by its effectiveness in GAPDH knockdown and GFP mRNA expression in human BM-MSCs. Together, our results demonstrated that AmCTOC liposomes showed superior transfection properties compared to commercial transfection agents, particularly in the 3D condition.
The selection of a suitable scaffold is also vital for the effective delivery of miRNA. One important criterion that we observed was using a scaffold with a neutral charge, since a charged scaffold impedes the nucleic acid release and interferes with the transfection process.28,29 In this study, charge-neutral collagen hydrogel was used for miRNA’s sustained delivery. It has already been used for cartilage regeneration in preclinical and clinical studies. Furthermore, it is well-known for biocompatibility, biodegradability, and low immunogenicity. 30 The collagen hydrogel was useful in the sustained release of growth factors, siRNA, miRNA, and drugs in other studies.29,31,32 However, the release kinetics of the collagen hydrogel showed a rapid release of about 90% of the encapsulated siRNAs was released within two weeks. An in vitro chondrogenic differentiation of MSCs takes a minimum of 3 to 4 weeks; a scaffold with an extended releasing profile (>3 weeks) would be ideal for cartilage tissue engineering.
Finally, our experiments established that the miR-140 activated collagen hydrogel using AmCTOC was successful in cartilage tissue engineering. The miR-140 is known to promote the chondrogenesis of MSCs. 4 In this study, we also demonstrated its ability to inhibit hypertrophy by showing its effect on decreasing the collagen type 10 gene and protein expression. The improved outcomes confirmed that the miR-140 sustained delivery through the activated matrix using a new 3D transfection agent produced stable hyaline cartilage-like tissue in vitro.
Collectively, this study provides in vitro proof for the usefulness of miRNA activated matrices for the preparation of hyaline cartilage from MSCs. Further preclinical testing in scaffolds with better mechanical and release characteristics is warranted to translate them into clinics. Another shortcoming of this study is that we used GFP expressing plasmid for screening the transfection reagents because we lacked access to the destabilized GFP (deGFP) expressing cell line. The deGFP cell line expresses a short half-life (≈2 hours) GFP protein, and it is commonly used for the optimization of RNA interference in 3D condition.8,33
Conclusion
This study found an optimal in vitro culture method to transfect MSCs in 3D conditions and demonstrated sustained delivery of miR-140 using an activated matrix for improved MSCs chondrogenesis with a lesser hypertrophic differentiation. Essentially, we identified a liposome that transfects cells in 3D condition with high efficiency. The developed strategy’s main advantage is minimal manipulation of cells ex vivo and providing cues for the transplanted or endogenous cells into the appropriate lineage. The findings pave the way for an off-the-shelf therapy for cartilage tissue engineering.
Supplemental Material
sj-pdf-1-car-10.1177_19476035211047627 – Supplemental material for Controlled Differentiation of Mesenchymal Stem Cells into Hyaline Cartilage in miR-140-Activated Collagen Hydrogel
Supplemental material, sj-pdf-1-car-10.1177_19476035211047627 for Controlled Differentiation of Mesenchymal Stem Cells into Hyaline Cartilage in miR-140-Activated Collagen Hydrogel by Karthikeyan Rajagopal, Porkizhi Arjunan, Srujan Marepally and Vrisha Madhuri in CARTILAGE
Supplemental Material
sj-pdf-2-car-10.1177_19476035211047627 – Supplemental material for Controlled Differentiation of Mesenchymal Stem Cells into Hyaline Cartilage in miR-140-Activated Collagen Hydrogel
Supplemental material, sj-pdf-2-car-10.1177_19476035211047627 for Controlled Differentiation of Mesenchymal Stem Cells into Hyaline Cartilage in miR-140-Activated Collagen Hydrogel by Karthikeyan Rajagopal, Porkizhi Arjunan, Srujan Marepally and Vrisha Madhuri in CARTILAGE
Footnotes
Supplemental Material
Acknowledgments and Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Science and Engineering Research Board (Principal Investigator: VM, Grant Number EMR/2015/000907), Department of Science and Technology, Government of India. Liposomal optimization was supported by the Department of Biotechnology (Principal Investigator: SM, Grant Number BT/PR25841/GET/119/162/2017), Government of India.
Declaration of Conflicting Interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
This study was approved by the Institutional Review Board and Ethics Committee (IRB Mins No. 9359), Christian Medical College, Vellore.
Informed Consent
Informed written consent was obtained before collecting the patient samples.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.