
Abstract
One of the most pressing issues in osteoarthritis (OA) research is the development of disease-modifying OA drugs (DMOADs), as currently there are no such drugs available. The paucity of suitable DMOADs is mostly due to the lack of approved ideal therapeutic targets necessary for the development of these drugs. However, based on recent discoveries from our laboratory and other independent laboratories, it is indicated that a cell surface receptor tyrosine kinase for collagen type II, discoidin domain receptor 2 (DDR2), may be an ideal therapeutic target for the development of DMOADs. In this article, we review the current status of research in understanding roles of DDR2 in the development of OA.
Keywords
Introduction
More than 30 million people are currently afflicted with osteoarthritis (OA) in the United States of America. 1 However, no disease-modifying OA drugs (DMOADs) are available to treat the disease. Existing drugs for the treatment of OA primarily release the pain and inflammation associated with advanced stages of the disease. Unfortunately, no amount of analgesics can prevent the cartilage from being destroyed. One of the unique characteristics in the development of OA is the long delay (spanning decades) between an initiating event and the onset of the pain, lack of mobility, and reduced quality of life associated with advanced OA. This provides us with the opportunity to search for novel therapeutic targets for the development of DMOADs.
Osteoarthritis is considered a whole joint disease that involves articular cartilage, subchondral bone, synovial membrane, menisci, and ligaments. 2 Abnormalities in any of the joint tissues, such as defective articular cartilage, alteration of subchondral bone remodeling, damage of joint ligaments, menisci and muscles, and synovitis can initiate or accelerate the pathological process, articular cartilage degeneration, which eventually leads to the destruction of articular cartilage. Regardless of the complexity of the OA etiology, the pathological progress of the articular cartilage degeneration is consistently characterized by the increased activity of chondrocytes at the early stages of the cartilage degeneration, followed by the degradation of proteoglycans and collagen type II, the appearance of fibrillation, and missing cartilage.3-6 This consistent pathological change of the cartilage degeneration suggests that there may be multiple initiating events toward the common targets that contribute to the cartilage degeneration. Therefore, the identification of the targets pathways may provide us with novel therapeutic targets for the design of clinical protocols in the prevention and treatment of OA.
Since the major cellular events in articular cartilage degeneration are the breakdown of collagen type II, and proteoglycans, much effort has been made to identify an enzyme(s) that can digest collagen type II and/or proteoglycans in the past 20 to 30 years. Matrix metalloproteinase-13 (MMP-13) is particularly important in the development of OA because of its ability to degrade both collagen type II and proteoglycans.7-9 Data from many investigations demonstrate that the expression of MMP13 is hardly detected in normal mature articular cartilages, but the activity and expression of the enzyme are increased in human OA cartilages and in mouse models of OA.10-13 The constitutive expression of MMP-13 in mouse cartilage results in OA-like changes to the knee joints, 14 and removal of this enzyme prevents articular cartilage erosion in a joint instability mouse model of OA. 15 The significant effect of MMP-13 in the development of OA is also highlighted by several other studies. A study by Verzijl et al. 16 indicates that the half-life of articular cartilage collagen in humans is 117 years. This slow rate collagen turnover suggests that chondrocytes may possess limited potential for collagen replenishment once the collagen is degraded within mature articular cartilage. Results from another study indicate that articular cartilage degradation is completely irreversible after the induction of MMP-mediated degradation of collagen type II and aggrecan. 17 If the degradation of articular cartilage by MMPs is irreversible, then we have to inhibit the activity and expression of MMPs in order to intervene in the progression of OA. In fact, numerous pharmacological companies have attempted to inhibit the activity of MMP-13 as a means of delaying the progression of OA. However, the broad biological effects of MMP-13 restrain its application as a target enzyme of inhibitor drugs in the treatment of OA. 18 For example, one significant unwanted effect of MMP inhibitors, including MMP-13 inhibitors, is the occurrence of musculoskeletal syndrome, a painful joint stiffening musculoskeletal symptom, and adhesive capsulitis (e.g., frozen shoulder). However, this concern does not apply to targeting molecules that induce the expression of MMP-13 in chondrocytes since those molecules may be more specific to chondrocytes.
Discoidin Domain Receptors as Candidate Cell Surface Signaling Molecules in Induction of MMP-13 in Chondrocytes
What led us to discover the role of discoidin domain receptor 2 (DDR2) in accelerating the process of articular cartilage degeneration, eventually leading to OA? Degradation of collagen type II is one of the major cellular events in the development of OA. MMP-13 is one of the major enzymes that degrade collagen type II. Data from a study indicates that MMP-13 is, at least, 10 times more efficient than MMP-3 in degrading collagen type II (8). This reminds us of a common cellular event in biological systems, enzyme induction by its own substrates ( Fig. 1 ).
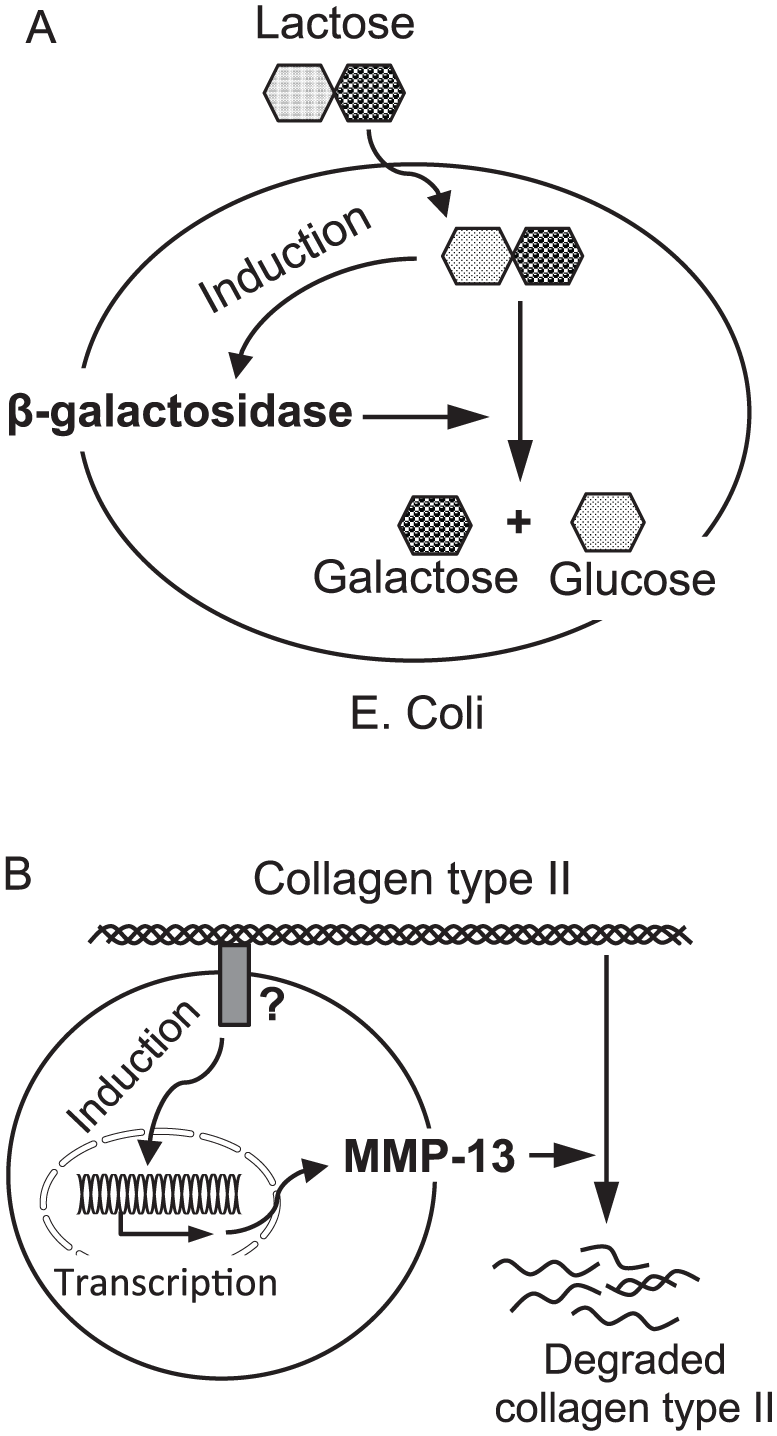
Enzyme induction by its own substrates. (
For cartilage turnover and remodeling, extracellular matrix molecules such as collagen type II need to be degraded, primarily by MMP-13. However, collagen type II cannot enter chondrocytes to directly induce MMP-13. Thus, the collagen has to interact with a cell surface receptor to activate intracellular signaling pathways to induce the expression of MMP-13 and the subsequent release of the enzyme into the extracellular space. If this is the case, the question becomes which cell surface molecule(s) transduce the signal into chondrocytes? We speculated that DDRs might be such a receptor.19,20 We performed immunohistostaining to examine expression profiles of DDR1 and DDR2 in human osteoarthritic cartilages from remaining sites of the femur head surface (surgical waste from hip replacements due to clinical OA).
We found that the protein expression of DDR2 was increased, whereas the protein expression of DDR1 was not detected in human osteoarthritic cartilages ( Fig. 2 ). We did not detect the protein expression of both DDRs 1 and 2 in the non-symptomatic human osteoarthritic cartilages.21,22 The result indicated that DDR2 might be a potential cell surface receptor that transduces a signal into chondrocytes to induce MMP-13. Results from our follow-up investigations (see the following discussions) suggest that DDR2 is a cell surface receptor that monitors the state of the extracellular matrix and can induce expression of MMP-13 in chondrocytes in response to cartilage damage. Data from 4 other independent studies support our observations. A study by Klatt et al. reports that interleukin (IL)-6, IL-8, and NFκB are the downstream effectors of the activation of DDR2 in induction of MMP-13 in chondrocytes.23,24 The result from an investigation by Vonk et al. demonstrates that a protein kinase C-dependent pathway is responsible for the upregulated expression of DDR2 itself in chondrocytes cultured on collagen type II–coated plates. 25 Holt et al. report that the increase in the expression of DDR2 is associated with the acceleration of articular cartilage degeneration in a spontaneous mouse model of OA. 26 An investigation by Suutre et al. indicates that the expression of DDR2 is increased in human OA cartilages. 27
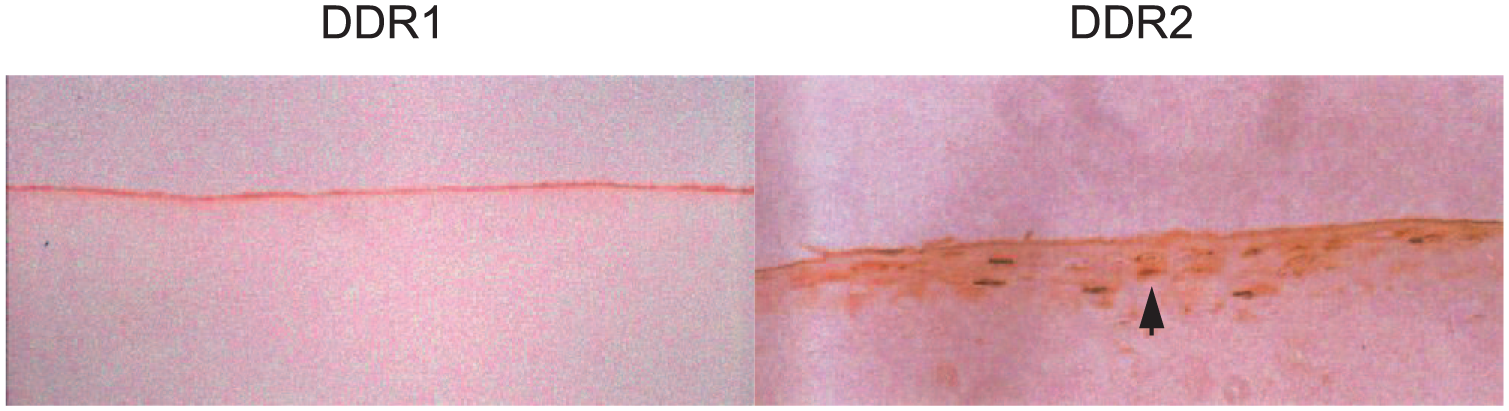
Immunohistochemistry staining of DDR1 and DDR2 in human degenerative articular cartilage. The protein expression of DDR2 was detected in degenerative articular cartilage (arrow; see brown color cells in the picture on the right), whereas the protein expression of DDR1 was undetectable in the cartilage (the picture on the left).
Induction of MMP-13 in Chondrocytes by the Interaction of DDR2 with Collagen Type II
In our investigations, we first examined whether or not integrin α2β1 was the cell surface receptor responsible for the transduction. 21 However, results from our study indicated that the blockage of the integrin α2β1 did not prevent the induction of MMP-13 in chondrocytes by collagen type II. This suggests that there is another cell surface receptor responsible for transducing the signal to induce MMP-13 expression in chondrocytes. We then examined whether or not DDR2 was responsible for the induction of MMP-13 in chondrocytes. We found that the expression of MMP-13 and DDR2 was increased in chondrocytes cultured on collagen type II–coated plates. 28 To confirm this observation, we overexpressed several forms of DDR2 in chondrocytes, including wild-type DDR2, DDR2 lacking the discoidin (DS) domain (ΔDS-DDR2), deletion of the protein tyrosine kinase (PTK) core (ΔPTK-DDR2), and a substitution of tyrosine for alanine at position 740 (Y740A-DDR2). We found that the overexpression of DDR2 increased the expression of MMP-13 in chondrocytes cultured on collagen type II–coated plates. The overexpression of ΔPTK-DDR2 or Y740A-DDR2 inhibited the elevated expression of MMP-13 and DDR2 in chondrocytes cultured on collagen type II–coated plates. Overexpressing ΔDS-DDR2 did not increase the expression of MMP-13 and DDR2 in chondrocytes cultured on collagen type II–coated plates. These results suggest that the interaction of DDR2 with collagen type II induces the expression of MMP-13 and upregulates expression of the receptor itself in chondrocytes in vitro. More important, our observation has been confirmed by 2 other independent research groups.23,24
We also investigated which signaling pathways were involved in the induction of MMP-13 in chondrocytes by the interaction of DDR2 with collagen type II. We cultured chondrocytes on collagen type II–coated plates with several chemical inhibitors, including MEK inhibitor PD98059, PI3 kinase/Akt inhibitor Wortmannin, JAK2 inhibitor AG490, p38 inhibitor SB203580, and JNKs inhibitor SP600125. We found that the inhibition of ERK and p38 signaling pathways inhibited the induction of MMP-13 in chondrocytes. The involvement of ERK pathway in the induction of MMP-13 was further confirmed by reduction of MMP-13 in chondrocytes overexpressing Raf kinase inhibitor protein in chondrocytes. 21 In addition, we found that the induction of MMP-13 by the activation of DDR-2 signaling was independent of the induction of MMP-13 by IL-1 signaling in chondrocytes. 28
In summary, the interaction of DDR2 with collagen type II activates the receptor and the downstream signaling pathways ERK and p38 to induce the expression of MMP-13 and a protein kinase C–dependent pathway to upregulate the expression of DDR2 in chondrocytes in vitro. It is now important to understand what the role of DDR2 is in the development of OA in vivo.
Attenuation of the Progressive Process of Articular Cartilage Degeneration by the Genetic Deletion of Ddr2 in Mice
To understand possible roles of DDR2 in the development of OA, we examined the expression profile of Ddr2 in joint cartilages of several mouse models of OA. Our studies demonstrated that collagen type IX deficiency (Col9a1−/−) and collagen type XI-haploinsufficiency (Col11a1+/−) in mice caused early onset articular and condylar cartilages degeneration.4,29 We also established 2 injurious mouse models of OA in our laboratory, a partial discectomy (PDE) of the disc in temporomandibular joint (TMJ) 5 and destabilization of the medial meniscus (DMM) in knee joint. 30 We found that expression of DDR2 was increased in the knee and TM joints of these mouse models of OA. We did not detect expression of DDR2 in normal joint cartilages. Moreover, the increase in expression of DDR2 was colocalized with elevated expression of MMP-13 in the joints of OA mouse models. Observation of upregulated expression of DDR2 in knee joints of an OA mouse model was confirmed by another independent research group. 26 In one of our follow-up investigations, we also found overexpression of DDR2 in human osteoarthritic knee joints, 22 which is consistent with results from another independent research group. 27
Upregulation expression of DDR2 was associated with human and mouse osteoarthritic cartilages. A question remained whether or not the increase in expression of DDR2 alone could initiate articular cartilage degeneration. To address this question, we created transgenic mice that conditionally overexpressed DDR2 in mature mouse articular cartilages, using the Tet-Off-inducible system. 31 The condition of articular cartilages from knee joints of transgenic mice was characterized. We also subjected the transgenic mice to DMM surgery. We found that the protein expression of DDR2 was increased in the knee joints of transgenic mice. However, the increased DDR2 did not induce MMP-13. We did not observe OA-like changes in the transgenic mice; however, when transgenic mice were subjected to DMM, the accelerated progression to OA was associated with DDR2 activation. These results suggest that conditionally overexpressed DDR2 cannot initiate articular cartilage degeneration in mature articular cartilage of mouse knee joints without activation of the receptor. Activation of the upregulated DDR2 can accelerate the progressive process of articular cartilage degeneration.
To address whether or not DDR2 could be an ideal therapeutic target for the development of DMOADs, we deleted Ddr2 from articular cartilages of mouse knee joints. Initially, we used Ddr2-deficient mice, generated by the conventional knockout techniques to removed 50% of Ddr2 in mice (Ddr2+/−). We then performed DMM on knee joints of Ddr2+/− mice. We also performed PDE on TMJ of Ddr2+/− mice. We found that the degenerative processes in articular and condylar cartilages, induced by either DMM or PDE, were significantly delayed in Ddr2+/−. We also genetically removed one copy of Ddr2 in a mouse model of OA (Col11a1+/−). We found that the degenerative processes of articular and condylar cartilages were significantly delayed in the Col11a1+/−;Ddr2+/− mice. 30 To provide further experimental evidence that DDR2 may be an ideal target for DMOADs, we conditionally removed Ddr2 specifically from cartilages in adult mice. 32 Ddr2 was removed at 2 time points: prior to and after the onset of articular cartilage degeneration induced by DMM. We found that the progressive process of articular cartilage degeneration, induced by DMM at both time points, was significantly delayed in the knee joints of Ddr2-deficient mice in comparison to their control littermates. These data suggest that DDR2 is likely one of the rate-limiting molecules in progressive joint failure, and that DDR2 may be an ideal target for the development of DMOADs. As we previously mentioned, one issue in the development of DMOADs is off-target effects of a drug. We found that the expression of DDR2 is not widely expressed in adult mouse tissues, only limited to the renal pelvis and testis. Another interesting point is that the conditional (local) removal of Ddr2 in the articular cartilage of adult mouse knee joints can protect articular cartilage from being degraded. This indicates that intraarticular injection of a biological reagent that inhibits the activation of DDR2 could have an adequate protective effect on joints against the development of OA.
Biological Functions of DDR2
Biological functions of DDR2 in normal and disease conditions have been intensively reviewed in 2 recent articles.33,34 Here, we briefly discuss biological roles of DDR2. (1) DDR2 can regulate multiple matrix metalloproteinases in cells, such as chondrocytes, human vascular smooth muscle cells, 35 human synovial fibroblasts, 36 human neutrophils, 37 rat hepatic stellate cells and murine skin fibroblasts.38-41 (2) DDR2 is required for the endochondral ossification during long bone growth. The genetic deletion of Ddr2 in mice (Ddr2−/−) by the conventional knockout technique results in a dwarfism phenotype, characterized by thinner growth plate, short long bones and shorter snout, due to the reduction in chondrocyte proliferation. 42 A spontaneous mutant mouse strain (slie), due to a loss-of-function mutation in Ddr2, shows a similar dwarfism phenotype observed in Ddr2−/− mice. 43 Loss-of-function mutations in DDR2 also cause a rare human genetic disorder, spondylo-meta-epiphyseal dysplasia with short limbs and abnormal calcifications.44-46 (3) DDR2 may play important roles in bone development by controlling osteoblast differentiation via phosphorylation of Runx2.47,48 In addition, the interaction of DDR2 with type I collagen induces lysyl oxidase in osteoblasts, 49 which promotes cross-linking of type I collagen fibers.
In Search of Small-Molecule Inhibitors of DDR2
One of the effective ways to inhibit induction of MMP-13 by the activation of DDR2 signaling in chondrocytes is to have small-molecule inhibitors that sufficiently inhibit the kinase activity of DDR2. Several DDR1 and 2 kinase inhibitors have been identified, such as imatinib, nilotinib, and dasatinib.50-52 However, those molecules are not DDRs-selective inhibitors and display the inhibitory effort on many other kinases. Thus, much effort is needed to identify novel small molecule inhibitors that specifically target DDR2. A very recent report by Grither and Longmore indicates that a small-molecule inhibitor, WRG-28, can specifically target the extracellular domain of DDR2 to inhibit the receptor signaling. 53 This provides us with an excellent opportunity to identify and develop highly selective DDR2 inhibitors as DMOADs. With regard to drug delivery, intraarticular injection of a small-molecule inhibitor can be one of the pharmacological treatments for OA, particularly in the consideration of the characteristic of the locally affected joints in the disease. By the intraarticular injection, we can minimize systematical off-target effects of drugs. Geiger et al. report that the application of cartilage-penetrating nanocarriers can improve the efficacy of growth factor treatment of OA in animal models. 54 This improvement of the local delivery of drugs can help us to discover DMOADs.
Summary
Based on the fact that the expression of DDR2 is elevated in human OA cartilages and the activation of DDR2 induces the expression of MMP-13 in chondrocytes and the genetic inactivation of Ddr2 can protect joints against the development of OA in animal models, DDR2 may be a potential target for the development of DMOADs.
Footnotes
Acknowledgments and Funding
This work was supported by NIH Grants R01-AR-051989 and Laboratoires Servier (Surensnes, France) research grant (to LX and YL).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.