
Abstract
Objective
Articular cartilage in mammals has limited intrinsic capacity to repair structural defects, a fact that contributes to the chronic and progressive nature of osteoarthritis. In contrast, Mexican axolotl salamanders have demonstrated the remarkable ability to spontaneously and completely repair large joint cartilage lesions, a healing process that involves interzone cells in the intraarticular space. Furthermore, interzone tissue transplanted into skeletal defects in the axolotl salamander demonstrates a multi-differentiation potential. Cellular and molecular mechanisms of this repair process remain unclear. The objective of this study was to examine whether paracrine mitogenic signals are an important variable in the interaction between interzone cells and the skeletal microenvironment.
Design
The paracrine regulation of the proliferation of equine interzone cells was evaluated in an in vitro co-culture system. Cell viability and proliferation were measured in equine fetal interzone cells after exposure to conditioned medium from skeletal and nonskeletal primary cell lines. Steady-state expression was determined for genes encoding 37 putative mitogens secreted by cells that generated the conditioned medium.
Results
All experimental groups of conditioned media elicited a mitogenic response in interzone cells. Fetal anlage chondrocytes (P < 0.0001) and dermal fibroblasts (P < 0.0001) conditioned medium showed a significantly higher mitogenic potential compared with interzone cells. Conditioned medium from bone marrow–derived cells elicited a significantly higher proliferative response relative to that from young adult articular chondrocytes (P < 0.0001) or dermal fibroblasts (P < 0.0001). Sixteen genes had expression patterns consistent with the functional proliferation assays.
Conclusions
The results indicate a mitogenic effect of skeletal paracrine signals on interzone cells.
Introduction
Articular cartilage in mammals has a limited intrinsic capacity to repair structural injuries and defects, a fact that contributes to the chronic and progressive nature of osteoarthritis. In contrast to mammals, Mexican axolotl salamanders (Ambystoma mexicanum) have demonstrated the remarkable ability to spontaneously and completely repair even large joint cartilage lesions, an intrinsic healing process that involves interzone cells in the intraarticular space. 1 Based on histological assessments of the repair process, interzone cells proliferate and extend into the lesion generating a primary repair tissue that then differentiates to restore normal tissue structures. An additional assessment of interzone tissue’s differentiation potential in axolotls was achieved through grafting experiments into critical sized defects created surgically in the tibia diaphysis. 2 Interzone tissue achieved closure of the defect, but rather than yielding diaphyseal bone, an entirely new joint was formed de novo complete with apposing articular surfaces and intervening interzone. In contrast, skin and muscle tissue transplants failed to bridge the skeletal defect in the tibia diaphysis. The repair tissue that bridged the bone gap in these studies matured to an anatomically organized structure resembling an intact joint with an interzone layer positioned in between two cartilaginous zones of tissue. The newly formed cartilage included cells with morphological features very similar to both epiphyseal/articular and metaphyseal chondrocytes. Cell tracking analysis by green fluorescent protein immunostaining confirmed that cells within the new diarthrodial joint were indeed derived from the green fluorescent protein transgenic donor interzone cells.
The studies outlined above suggest a model whereby interzone cells respond to paracrine inductive signals from the skeletal environment to proliferate and spontaneously differentiate into multiple synovial joint tissues within the bone defect. Important issues that remain unknown, however, include the cellular and molecular mechanisms regulating this repair process. Are interzone cells autonomously regulated, directing their own proliferation and differentiation to form articular cartilage and other synovial joint tissues? Or, are interzone cells regulated by neighboring cells, mechanisms that might be niche-dependent and sensitive to different tissue microenvironments? What are the specific signaling pathways that regulate the interzone-mediated cartilage repair process and de novo neo joint formation observed in axolotl salamanders? Interestingly, the cells in the repair tissue do not proliferate beyond the defect suggesting a regulatory control over cell proliferation. The primary focus of this study was to investigate paracrine regulation of interzone cell proliferation. Paracrine mechanisms that regulate interzone cells in a skeletal microenvironment may provide new insight into the potential of interzone for cell-based therapy for the repair of synovial joint defects involving the articular surface. Tissue culture studies with axolotl cells are very difficult due to technical challenges associated with maintaining amphibian cells in vitro. For this reason and due to likely targeted future patient populations, a mammalian in vitro co-culture model using equine primary cells was utilized to examine a potential paracrine interaction between skeletal cells and the interzone.
This study was designed to test the hypothesis that paracrine signals derived from skeletal cells stimulate interzone cell proliferation in an in vitro system employing conditioned medium as the source of paracrine mitotic stimuli. The specific objectives were to compare relative changes in viability and proliferation of equine fetal interzone cells in response to paracrine signals secreted by different primary skeletal and nonskeletal cells in conditioned medium. Signaling molecules such as FGFs and TGFs that are expressed during development have been shown in some systems to be re-expressed following tissue injury.3,4 As such, adult synovial joint tissues may have the potential to regulate interzone cell proliferation, migration, and differentiation in a repair context. Therefore, we examined an effect of donor age on the ability of cells to induce proliferation of interzone cells. We hypothesized that embryonic cells exhibit a higher proliferative potential compared to cells derived from older individuals. 5 In addition, a transcriptomic approach was employed to characterize the secretome of the cells that generated the conditioned media and to identify potential growth factors involved in the paracrine interaction.
Methods
Isolation and Culture of Cells
Primary cells were isolated from equine fetuses and young adults (peripubescent horses between 15 and 17 months of age), and expanded in culture as described previously. 6 Briefly, mares were bred by natural cover after estrous cycle synchronization and gestational age was confirmed through a transrectal ultrasound examination to determine embryonic vesicle size. 7 Fetuses were recovered intact at 45 or 46 days of gestation from mares (one fetus per mare) while standing and sedated, using a minimally invasive uterine lavage technique that displaced the embryo within its vesicle. The fetus was then recovered into a sterile bag by gravity evacuation of the lavage fluid back through the tube. Immediately after collection, fetuses were thoroughly rinsed in ice-cold sterile Dulbecco’s phosphate buffered saline (PBS; Gibco, Cat# 14190144) with 2% (v/v) amphotericin B (anti-mycotic, Gibco, Cat# 15290026) and 2% (v/v) penicillin/streptomycin (P/S; antibiotic; Gibco, Cat# 15070063). Each fetus was catalogued and transported in a sterile container on ice for immediate tissue processing. All procedures were conducted in accordance with a University of Kentucky Institutional Animal Care and Use Protocol (IACUC #2014-1215).
Interzone cells, anlage chondrocytes, and dermal fibroblasts were isolated from one forelimb and one hindlimb of each fetus as described previously. 6 Briefly, interzone cells were liberated from the cuboidal bone anlage of developing carpal and tarsal joints (Suppl. Fig. S2) by digestion in 500 µL of commercial 0.25% trypsin ethylenediaminetetraacetic acid solution (EDTA; Gibco, Cat# 25200056) for 5 to 10 minutes with gentle agitation using a micropipette. The enzymatic activity of trypsin was then quenched with 1 mL Dulbecco’s modified Eagles medium (DMEM; Gibco, Cat# 10569044) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS), 1% (v/v) P/S, and 1% amphotericin B (FBS medium). Undigested tissue fragments and cuboidal bone anlagen were allowed to settle by gravity for 3 minutes. The supernatant containing the suspended cells was then transferred to a sterile 15-mL polypropylene tube, and centrifuged at 1000 rpm for 3 minutes. The resulting pellet was resuspended in 5 mL of FBS medium and plated in 6-well cell culture plates (Bio-Star, VWR Cat# 10062-892). Anlage chondrocytes were isolated from internal cubes of tissue dissected from distal metaphyseal anlage of the developing humerus and femur with careful attention to avoid inclusion of the epiphysis, diaphysis, or anlage surface. To further minimize the potential for cell contamination, the anlage cubes were incubated in 500 µL of the commercial 0.25% trypsin EDTA solution for 5 to 10 minutes, followed by gravitational settling of the anlage cubes. This was followed by two rinses in sterile PBS supplemented with 2% (v/v) P/S and 2% (v/v) amphotericin B. Chondrocytes were then liberated from the matrix using 1 mL of 0.5% collagenase D (Worthington, Cat# CLS4) for 10 to 20 minutes with gentle agitation using a micropipette. Quenching of the collagenase enzyme activity, isolation, and plating of primary anlage chondrocytes were performed in a similar fashion as described above for interzone cells. Fibroblasts were isolated from the truncal dermis targeting a region of skin far removed from any skeletal structures. Care was taken during tissue dissection to ensure exclusion of epidermis and hypodermis. The subcutaneous dermal tissue was then finely minced, followed by digestion in 1 mL of the commercial 0.25% trypsin EDTA solution for 10 to 20 minutes with gentle agitation using a micropipette. Separation of undigested tissues, quenching of enzyme activity, isolation, and plating of primary dermal fibroblasts were similar to the procedures described above for interzone and anlage chondrocytes.
Articular chondrocytes, bone marrow–derived cells, and dermal fibroblasts were isolated from young adult horses as described previously. 6 Briefly, tissue samples were collected immediately postmortem from 5 young adult horses of mixed light breed heritage that were euthanized at 15 to 17 months of age for reasons unrelated to the current study (IACUC #00843A2005). Bone marrow aspirates were collected from the sternum using an 8 FrG Jamshidi needle and heparinized syringes. 8 The samples were stored on ice until processing, which involved direct plating in 1:1 FBS medium in T-75 tissue culture flasks (Bio-Star, VWR Cat# 82050-856). The cultures were rinsed with PBS over the course of the first few days of culture to facilitate removal of red blood cells and yield adherent cells. Dermal fibroblasts were isolated from the dermal layer of skin at the tail base. Dermis was minced and transferred to tissue culture plates in FBS medium. Resulting dermal tissue explants were then maintained in FBS medium for several days to yield adherent dermal fibroblasts. The processing of bone marrow and dermal tissue samples were completed within 3 hours post-mortem. Articular chondrocytes were isolated as previously described. 9 Briefly, articular cartilage of the femorotibial joint was shaved down to the calcified layer. Cartilage shavings were copiously rinsed in PBS with 2% antimycotic and antibiotic supplements as described above. The shavings were then minced, weighed, and digested for 22 hours in medium (OptiMEM [Gibco Cat # 51985091, 5% FBS, 1% antimycotic/antibiotic) containing bacterial collagenase (Worthington) at 7.5 mg of collagenase per gram of cartilage. Primary chondrocytes released from the cartilage matrix were centrifuged, rinsed, counted, and plated into T-75 tissue culture flasks at a seeding density of 2.1 × 106 cells per flask.
Taken together, a total of 6 primary cell lines isolated from 5 equine fetuses and 5 young adult horses were used for this study. Each biological replicate provided all 3 fetal or all 3 young adult cell lines, respectively.
Conditioned Medium Preparation
The 6 primary cell lines isolated from skeletal and nonskeletal tissues of equine fetuses (n = 5) and young adult horses (n = 5) were used individually for the preparation of conditioned medium. To prepare the conditioned medium, thawed P2 cells were passaged and plated at P3 into five T-75 tissue culture flasks at a seeding density of 500,000 cells per flask in FBS medium. Medium was replenished every 48 hours until the cells reached 70% to 80% confluence. The cells were then rinsed thrice with PBS, and incubated for 24 hours in 5 mL of DMEM supplemented with 1% (v/v) P/S, and 3 mg/mL of bovine serum albumin (BSA; Sigma Cat# A9418). This amount of BSA was calculated to be roughly equivalent to the amount of total protein in 10% FBS. The preparation is referred to as “BSA medium” is this article. In control experiments, 24 hours of conditioning was found to elicit a greater mitogenic response with young adult dermal fibroblasts in comparison to medium conditioned for 72 hours (Suppl. Fig. S3). At the end of the conditioning period, the media from the five T-75 flasks were pooled for each cell type and each biological replicate, centrifuged, steri-filtered using a 0.2 µM polyethersulfone membrane filter (VWR, Cat # 28145-501), and stored at −80°C in aliquots. For normalization, cells from each corresponding group of five T-75 flasks were trypsinized and counted, and the total cell count that generated the volume of conditioned medium was recorded and used for normalization throughout this study.
Co-Culture
To minimize background cell proliferation in the experimental system prior to assessing the mitogenic activity of the conditioned medium, a determination was made on growth arresting the P3 cells through FBS deprivation to the point where the rate of cell division approached zero, but cell viability remained high. This was determined empirically for each cell line studied. P3 cells were plated in FBS medium for 24 hours after which the complete medium was removed and the cells refed with BSA medium. The cells were maintained in this serum-deprived state, with viability and proliferation assessments performed as described below every 48 hours. BSA medium was also refed at 48-hour intervals. The functional ability of the serum starved cells to respond to mitogenic stimuli was then evaluated by refeeding with FBS medium.
Sample size was determined by power analysis. Power calculations were performed using data from a pilot study (Suppl. Table S2) to assess interzone cell proliferation in response to conditioned medium from fetal skeletal and nonskeletal cell lines (n = 2). The estimated sample size required to achieve a power of 0.80 was two.
Responder Cells to Test the Different Conditioned Medium Preparations
Interzone cells at P3 were plated in 24-well plates in FBS medium at a seeding density of 50,000 cells per well to yield an adherent monolayer. Following 24 hours of culture, the medium was replaced with BSA medium for 5 days to minimize the level of background proliferation. On day 5, the interzone cell monolayers were refed with either conditioned medium derived from one of the experimental cell lines, BSA medium as a negative control, or FBS medium as a positive control. The conditioned medium derived from fetal (n = 5) and young adult (n = 5) primary cell lines served as the source of mitogenic stimuli. Six technical replicates were performed for each conditioned medium group and 3 replicates each for positive and negative control media.
Differential Cellular Response to the Same Level of Mitogenic Stimuli
In addition, we investigated whether any of the cell types exhibit a differential response to the same level of mitogenic stimulation. The primary cell lines isolated from 5 equine fetuses (n = 5) and 5 young adult horses (n = 5) were evaluated independently as responders, while the mitogenic stimuli in fetal anlage chondrocytes-derived conditioned medium was held constant. Eight technical replicates were performed for all cell lines in conditioned medium and 4 in each control media. Plating and co-culture for all the cell lines were performed as described above.
Assessment of Cell Viability
Cell viability was assessed using a commercial LIVE/DEAD Viability/Cytotoxicity Kit for mammalian cells (ThermoFisher Scientific, Cat# L3224) according to the manufacturer’s protocol. Briefly, adherent cells in 24-well plates were washed twice with PBS to remove residual serum esterase activity. The cytoplasm of live cells stains with Calcein AM (1 µM), while nuclei of dead cells stain with ethidium homodimer-1 (2 µM). All cell nuclei were counterstained with Hoechst 3342 (5 mg/mL) (ThermoFisher Scientific, Cat# 3570). The total number of cell nuclei and the dead cell nuclei per field were counted using automated imaging software (NIS elements 4.0, Nikon Instruments Inc.) and the percent viability was calculated as the number of live cells per 100 cells total. The counts were recorded in 3 distinct fields and averaged for each well.
Assessment of Cell Proliferation
Levels of cell proliferation were quantified by measuring the incorporation of 5-ethynyl-2′-deoxyuridine (EdU) following a pulse labeling. EdU incorporation and subsequent visualization were performed as described previously. 10 Briefly, cells were labeled with a 24-hour pulse of EdU (Jena Bioscience, Cat# CLK N001-25) at a concentration of 8 µM. The cells were then fixed in 4% paraformaldehyde in PBS prior to permeabilization with 0.1% Triton-X followed by detection. For detection of the EdU label, the cells were incubated for 30 minutes in 100 mM Tris, 4 mM copper(II) sulfate (Sigma, Cat# 451657), 100 µM biotin conjugated azide in dimethyl sulfoxide (Jena Bioscience, Cat# CLK-AZ104P4-25), and 100 mM ascorbic acid (Sigma, Cat# 451657). The staining mix was prepared fresh for each assay and was used for staining cells immediately after the addition of ascorbic acid. The EdU label was visualized with streptavidin-conjugated Texas Red (Vector Biolabs, Cat# SA 5006). The cells were counterstained with DAPI (4′,6-diamidino-2-phenylindole; Life Technologies, Cat# D1306). The total number of cell nuclei and the number of proliferating cells (EdU labeled) were counted using automated imaging software (NIS elements 4.0, Nikon Instruments Inc.) by fluorescence microscopy. The rate of proliferation was calculated as the number of EdU-labeled cells as a percent of the total number of cells in each field. The percent proliferation was calculated for 3 fields per well and the values averaged.
Differential Gene Expression
To compare patterns of gene expression in the cell lines used to generate conditioned medium, RNA-seq data generated from P3 monolayer cultures of the same primary cell lines as part of a related study were utilized. 11 A list of 37 genes with established functional annotation related to the regulation of cell proliferation (Suppl. Table S1) was generated using Amigo 2.0. 12 The number of RNA-seq reads that uniquely aligned within the structural nucleotide coordinates for the Refseq mRNA for each gene locus were normalized by multiplying the read counts for each gene by a factor accounting for both sequencing depth of each individual library and length of the specific target gene. Comparisons of the normalized read counts were made on a gene-by-gene basis across all the biological replicates of the samples.
Statistics
Data from the viability and proliferation assays were independently evaluated by incomplete block analysis of variance using SAS statistical software, version 9.3 (SAS Institute Inc., Cary, NC). Pair-wise comparisons were made between the treatment means with a Bonferroni post hoc correction for multiple comparisons. Differences were considered statistically significant if the corrected P value was <0.005. Age of the donor from which primary cells were isolated was evaluated as an additional factor and statistical significance was defined at P < 0.05. Residual plots, histograms of residuals, and Q-Q plot of residuals were evaluated to ensure that the normality assumptions were not violated. Normalized read count data of differentially expressing genes from pairwise comparisons were evaluated using one-way analysis of variance using SAS statistical software, version 9.3, with a Tukey-Kramer post hoc correction for multiple comparisons (SAS Institute Inc., Cary, NC). Differences were considered statistically significant at P < 0.05.
Results
Background Proliferation
The amount of time in BSA medium to achieve a minimal level of background proliferation in culture was identified for each cell line empirically. They were as follows:

The cell lines were then evaluated after refeeding with FBS medium. In each case, the 48hour time point after refeeding exhibited the maximum percentage of proliferating cells, likely due to synchronization of the cell cycle. Cell viability was supported by refeeding with FBS medium, but remained above 94% even in cells maintained in BSA medium. Supplemental Figure S1 illustrates data from interzone cells, but is representative of all 6 cell lines.
Different Mitogenic Activity in Conditioned Medium
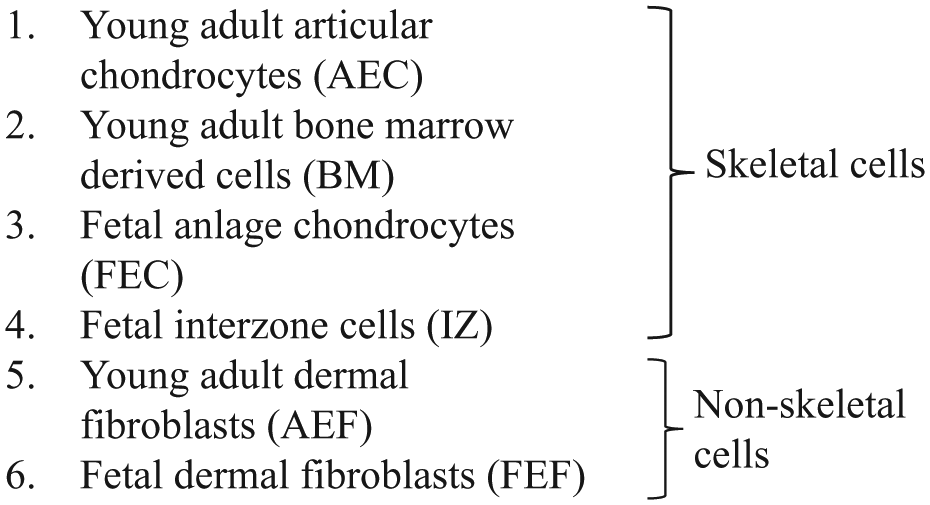
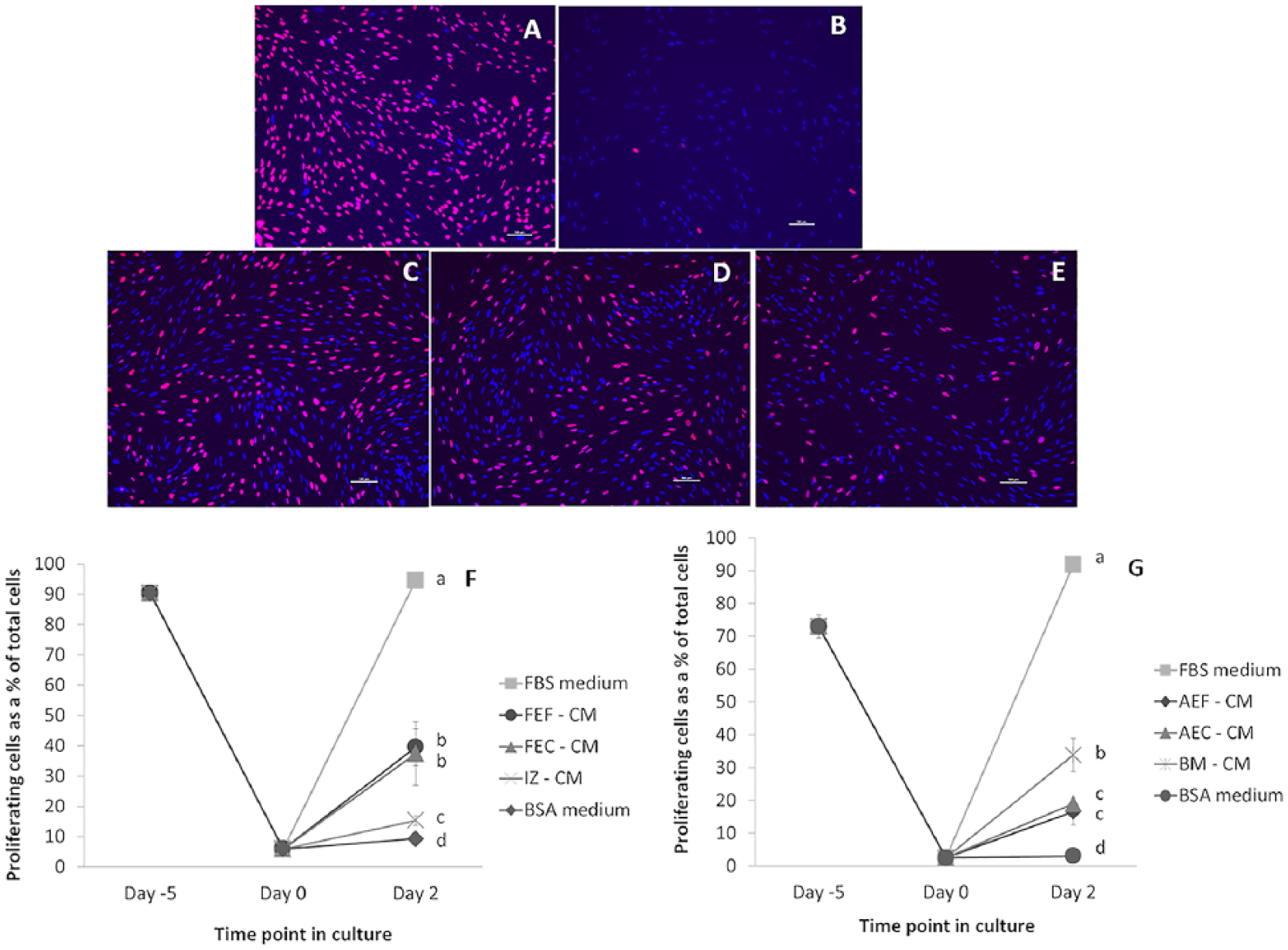
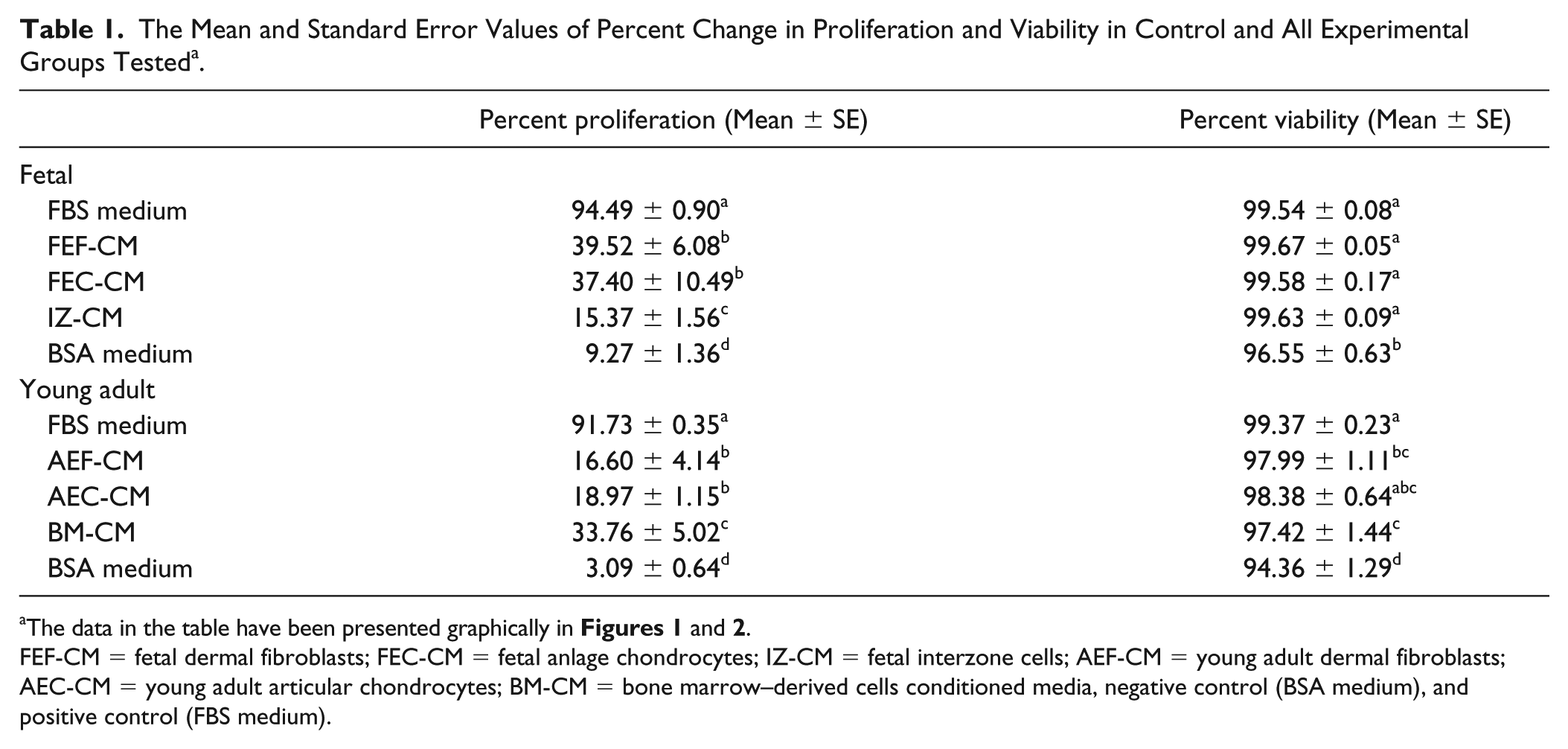
The level of proliferation was determined by measuring the number of cells that incorporated the EdU label as a percent of the total number of cells and has been presented in Figure 1 and Table 1 . Figure 1A-E presents representative images of proliferating interzone cells (48 hours post-treatment) in response to medium conditioned by the different fetal cell lines. Figure 1F illustrates the percent proliferation trend in the fetal cell conditioned media over the time course of the study, which was as follows: FBS medium (with a mean percent proliferation of 94 ± 0.9) > Fetal dermal fibroblast CM (39 ± 6.1) = Fetal anlage chondrocyte CM (37 ± 10.5) > Interzone cell CM (15 ± 1.5) > BSA medium (9 ± 1.4). The interzone cells in the positive control medium exhibited the highest percent proliferation, approaching 100% in all the experiments. In contrast, the interzone cells in the negative control group had the smallest number of positively stained cells and the lowest rate of proliferation. Based on a p-value threshold corrected for multiple comparisons, all conditioned media groups generated a significantly higher level of cell proliferation in interzone cells relative to the negative control medium. Medium conditioned by fetal anlage chondrocytes (FEC-CM) were significantly more mitogenic (P < 0.0001) compared to interzone cell conditioned medium (IZ-CM). There was no significant difference in the percent proliferation values of interzone cells in fetal dermal fibroblast (FEF-CM) and anlage chondrocyte conditioned medium. However, some variation was observed in the mitogenic induction ability within the FEC-CM group compared to other groups. Interestingly, IZ-CM was the least mitogenic among all the groups, but still stimulated cell proliferation at a level significantly higher than the negative control medium (P < 0.001).
(
The Mean and Standard Error Values of Percent Change in Proliferation and Viability in Control and All Experimental Groups Tested a .
FEF-CM = fetal dermal fibroblasts; FEC-CM = fetal anlage chondrocytes; IZ-CM = fetal interzone cells; AEF-CM = young adult dermal fibroblasts; AEC-CM = young adult articular chondrocytes; BM-CM = bone marrow–derived cells conditioned media, negative control (BSA medium), and positive control (FBS medium).
Interzone cells cultured with conditioned medium from all 3 types of young adult cells also exhibited significantly higher mitogenic activity than the negative control medium ( Fig. 1G ). Interzone cells in the positive control (92 ± 0.3) and negative control (3 ± 0.6) medium showed the maximum and minimum percent proliferation values, respectively. The bone marrow–derived cell conditioned medium (34 ± 5.0; BM-CM) showed significantly higher mitogenic potential compared with young adult chondrocyte (19 ± 1.1; AEC-CM; P < 0.0001) and young adult dermal fibroblast (17 ± 4.1; AEF-CM; P < 0.0001) conditioned medium. The difference in percent proliferation values of interzone cells in AEC-CM and AEF-CM did not reach statistical significance (P = 0.0883).
In addition, an effect of donor age on the mitogenic potential of skeletal and nonskeletal cells was evaluated. Conditioned media derived from fetal anlagen chondrocytes were significantly more mitogenic compared with those from young adult articular chondrocytes (P = 0.0035). Conditioned media derived from fetal fibroblasts elicited significantly higher mitogenic response relative to young adult fibroblasts (P = 0.0008).
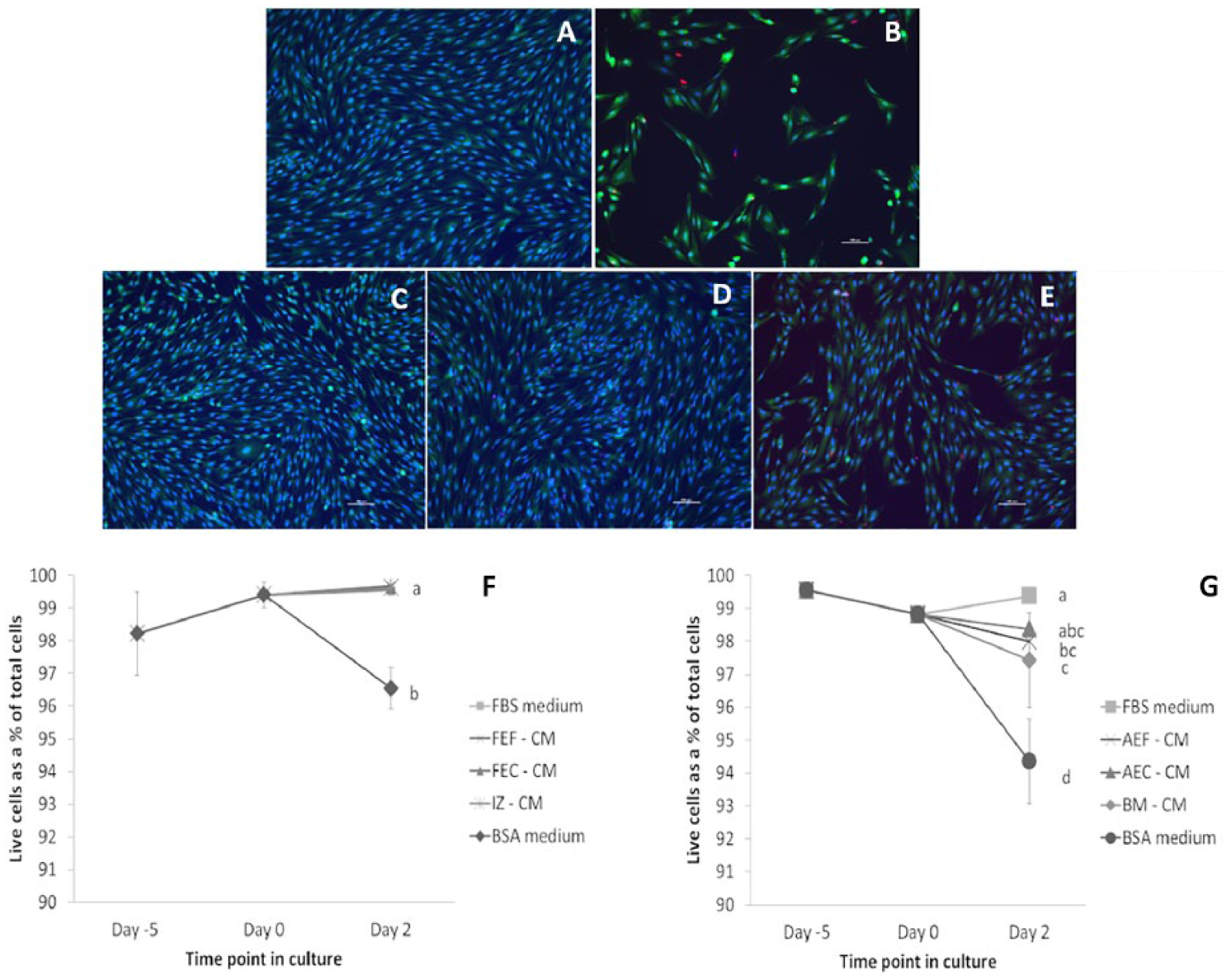
Cell viability was assessed by measuring the number of live cells as a percent of the total number of cells. Interzone cells in the different fetal cell conditioned media and positive control medium showed significantly greater viability (>99%) relative to those in the negative control serum-free medium, which still retained cell viability at more than 95% ( Fig. 2 and Table 1 ). Interzone cells in the young adult cell conditioned medium and positive control medium were also significantly more viable compared with the negative control medium. In the conditioned media groups, there was some variation in the percent viability. However, it is worth noting that the lowest percent viability observed in our study was 94%, indicating that a large majority of the cells remained viable within the time frame studied despite removal of FBS or another supplemental source of growth factors.
(
Differential Cellular Response to the Same Level of Mitogenic Stimuli
The 6 different cell lines demonstrated a positive but variable proliferative response when exposed to the same conditioned medium. Fetal dermal fibroblasts showed a significantly higher response compared with skeletal cell types. Fetal anlage chondrocytes showed a significantly lower percent proliferation relative to other fetal cell types
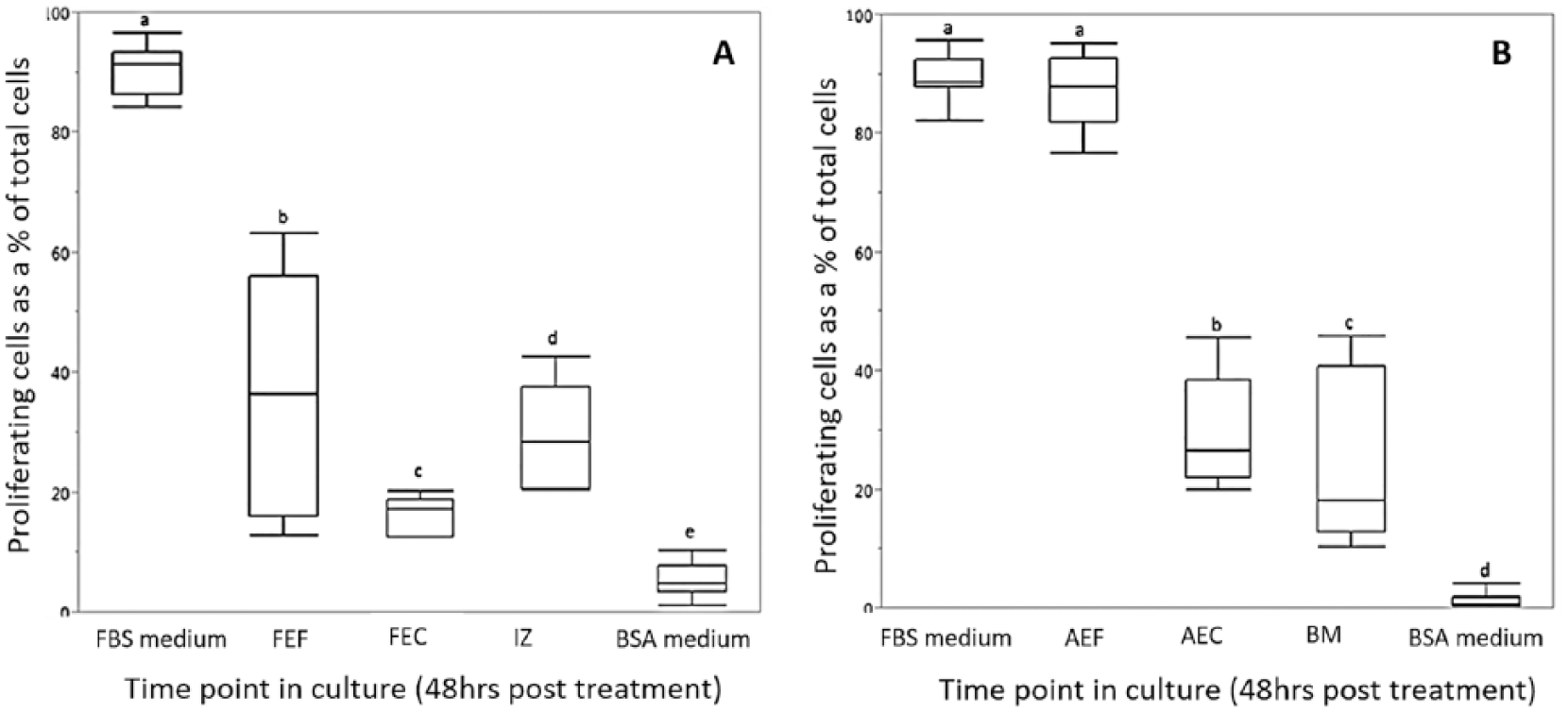
Box and whiskers plot representing the percent change in proliferation of fetal (
Differential Gene Expression
The average normalized RNA-seq read counts for all samples are presented in Table 2 . Analyses of differential gene expression based on the number of read alignments was used to generate P values for each gene-by-gene statistical test. Out of the 37 genes that were identified using Amigo 2.0 based on their functional annotation, the normalized read counts of 10 genes in the fetal group and 6 genes in the young adult group demonstrated a pattern of differences across the cell types consistent with that observed in the functional assays ( Tables 3 and 4 ). In the fetal group, IZ consistently showed the lowest read counts for all 10 genes; however, not all of these differences reached statistical significance. The read counts followed the trend observed in the proliferation functional assay for platelet-derived growth factor subunit D (PDGFD) with values in FEF (P = 0.0287) and FEC (P = 0.0156) being significantly higher than in IZ. Normalized read counts were significantly higher in FEC (P = 0.0096) for epithelial mitogen (EPGN), and in FEF (P = 0.0001) for platelet-derived growth factor subunit B (PDGFB) compared to IZ. In the young adult group, the read counts of placental growth factor (PGF) followed the pattern observed in the functional assays with values in BM being significantly higher than in AEF (P = 0.0035) and AEC (P = 0.0287), whereas the differences in the other genes were not statistically significant.
Normalized Gene Counts for the Targeted Genes with a Biological Function of Proliferation for Each Cell Type a .
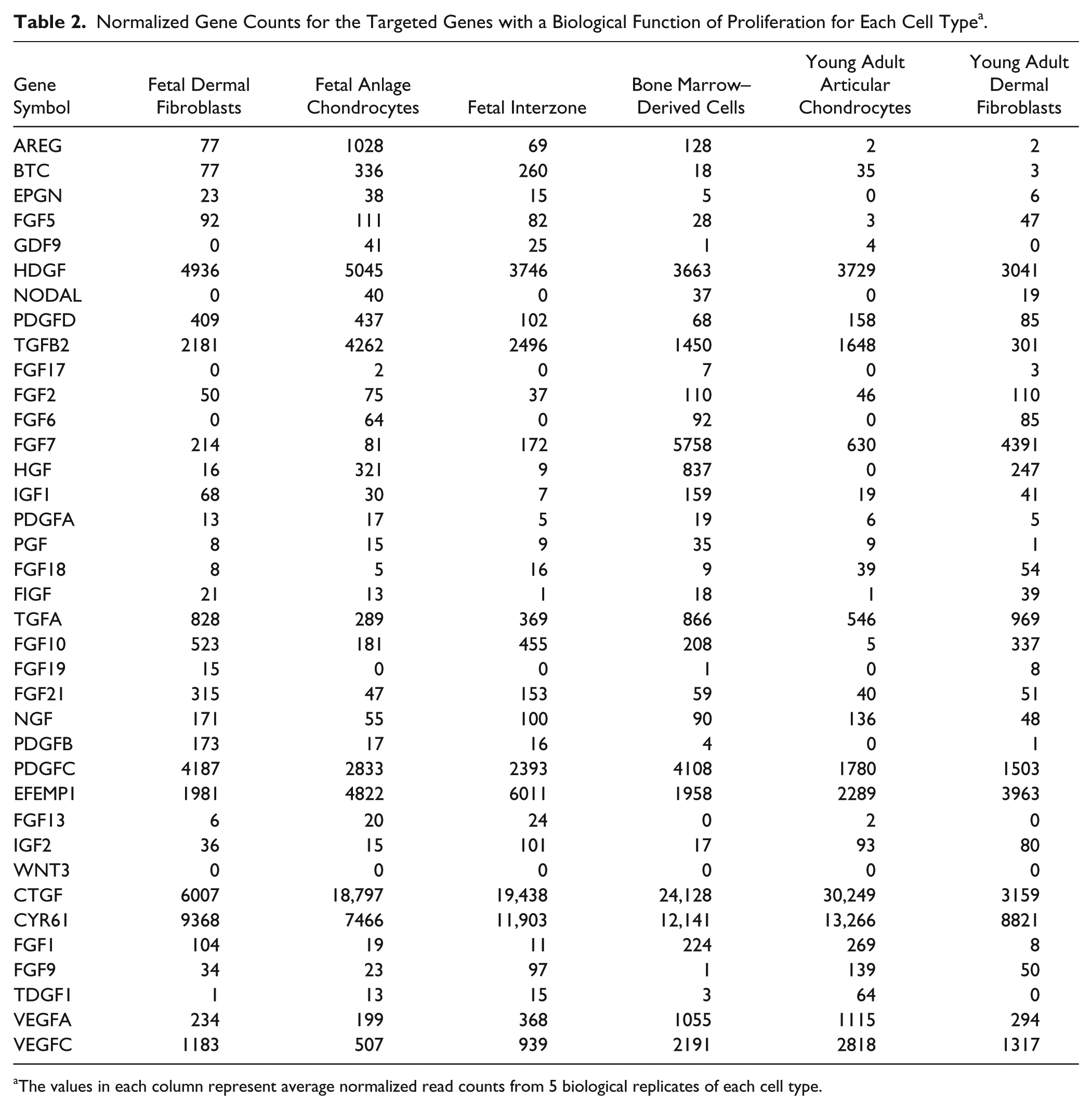
The values in each column represent average normalized read counts from 5 biological replicates of each cell type.
List of Genes for Which the Normalized Gene Counts for Fetal Cell Types Follow the Trend Observed in the Functional Assay*.
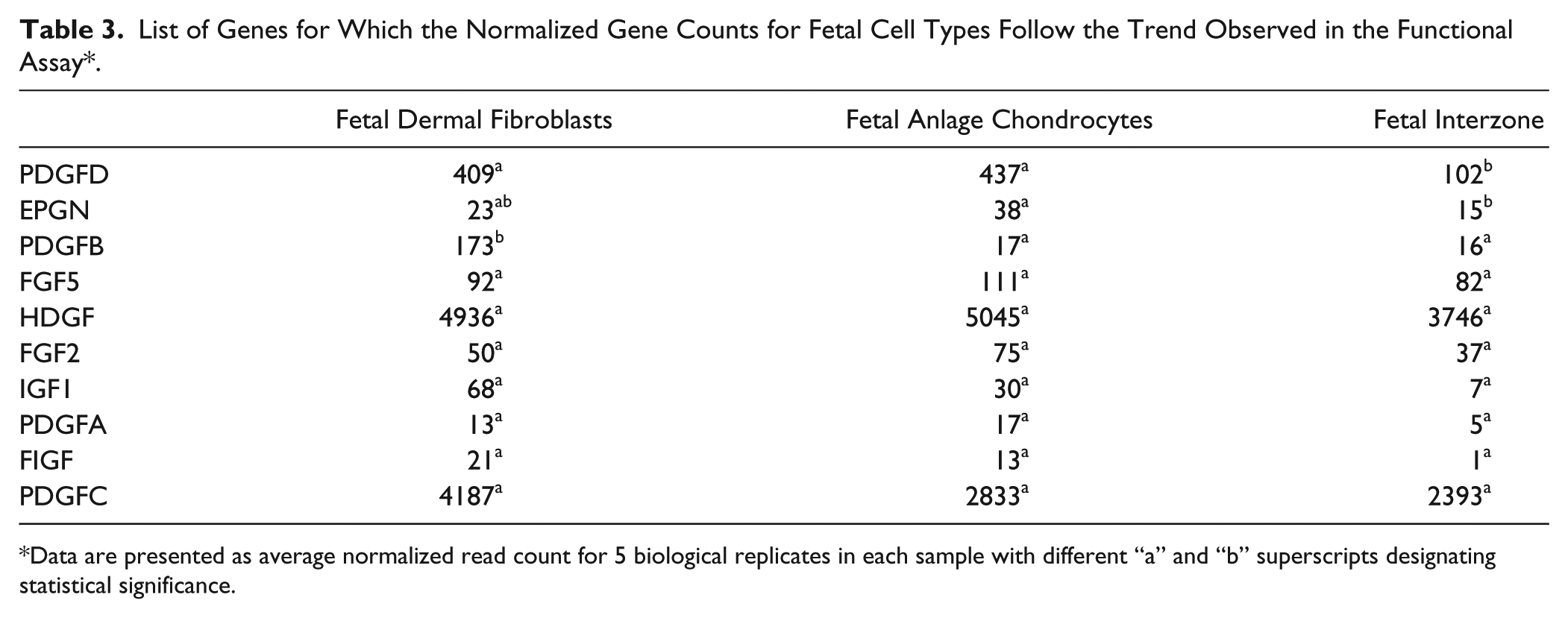
Data are presented as average normalized read count for 5 biological replicates in each sample with different “a” and “b” superscripts designating statistical significance.
List of Genes for Which the Normalized Gene Counts for Young Adult Cell Types Follow the Trend Observed in the Functional Assay*.
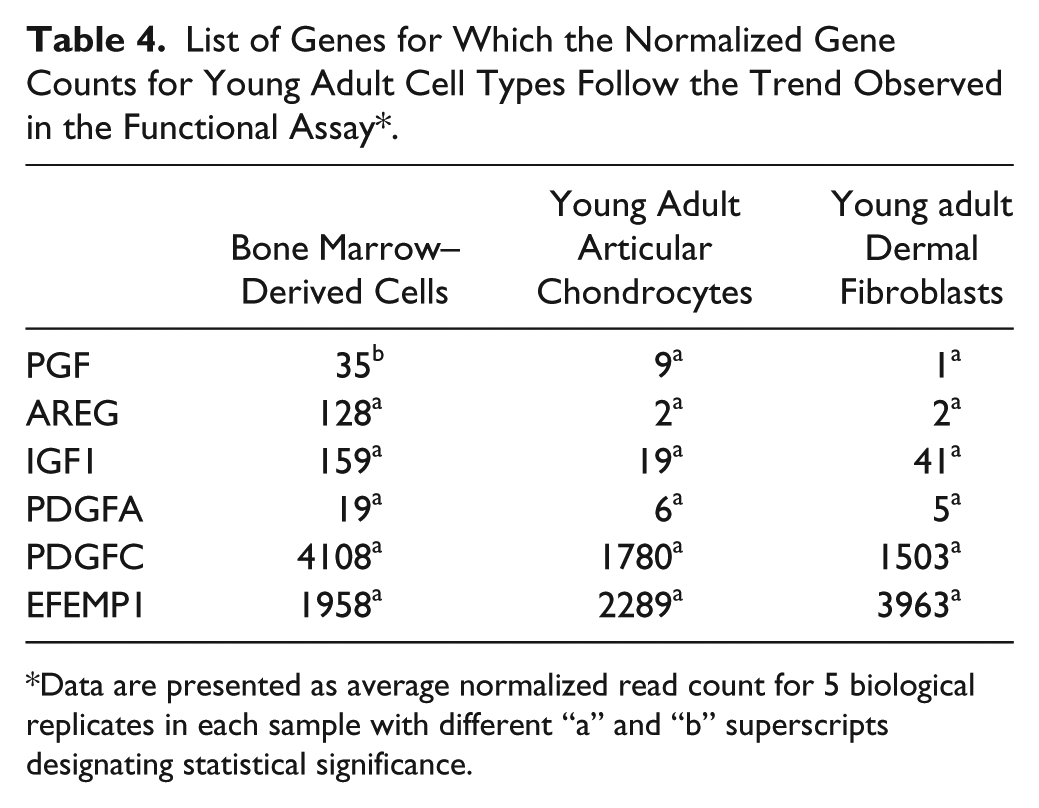
Data are presented as average normalized read count for 5 biological replicates in each sample with different “a” and “b” superscripts designating statistical significance.
Discussion
In this study, we examined how conditioned media, and thus, secreted paracrine signals from different cell lines, affected the proliferation and viability of interzone cells in co-culture. Interzone cells used in this study were at passage 3 and maintain the expression of 10 targeted genes between passages 3 and 7 in monolayer interzone cultures. 13 Since a change in the rate of proliferation could be a function of differences in the mitogenic stimuli that the cells are exposed to, as well as how the same stimulus is perceived by different cell types, we also investigated whether the skeletal and nonskeletal cell types would respond differentially to the same mitogenic stimuli. Additionally, we assessed donor age as a variable by comparing mitogenic responses in cells of fetal and young adult origin. The findings from this study support the hypothesis that skeletal cell–derived paracrine signals have a mitogenic effect on interzone cells.
All experimental groups of conditioned media elicited a significant mitogenic response in interzone cells, the amount falling between levels of stimulation induced by the positive and negative control media. In the fetal group, FEC-CM and FEF-CM showed a significantly higher mitogenic potential compared to IZ-CM. Among the young adult cell conditioned media, BM-CM stimulated significantly more cell proliferation than AEC-CM and AEF-CM. Comparisons between fetal and young adult cells were confounded by cell type differences, with dermal fibroblasts being the most analogous between the 2 ages. In general, though, the data indicate that conditioned media from fetal cells had higher mitogenic activity relative to young adult cells.
The conditioned medium system utilized in this study is a widely used indirect co-culture model for studying paracrine interactions between distinct cell populations.14-18 It provides several advantages over other co-culture models, including the ability to control the volume of conditioned medium produced and the length of the incubation period. We added cell counts from the tissue culture flasks after collection of conditioned medium for further control of technical variation. Additionally, the separate physical locations of cells producing conditioned medium and the responder cells reduces ambiguity of signaling directionality. We found this co-culture model to be useful for assessing the mitogenic potential of paracrine signals from all the cell types tested.
The data support a model that includes paracrine signaling between skeletal tissues and interzone cells in the developing joint. Further mechanistic studies will be valuable in determining the identity and signaling pathways of specific interzone cells regulators, several of which have already been implicated.19-21 It will be interesting to investigate whether these variables are also active in joint tissue repair processes.
In an effort to characterize the secretome of the cells studied and to identify potential mitogenic factors in the conditioned medium, RNA-seq-based expression profiling of 37 genes annotated to the term “regulation of cell proliferation” was performed across all samples. In the fetal group, 10 genes demonstrated expression patterns concordant with the results of the functional proliferation assays. Platelet-derived growth factors (PDGFs) are well-documented mitogens, driving the proliferation of undifferentiated mesenchymal cells during early development. In later stages, they play an important role in angiogenesis, tissue remodeling, and cell differentiation.22-24 PDGFA and PDGFB have been shown to stimulate the proliferation of human bone marrow-derived mesenchymal stem cells (MSCs). 25 In addition, PDGFA has been implicated in the formation of smooth muscle actin filaments in MSCs. 26 PDGFC and PDGFD have been shown in human MSCs to have a robust effect on the proliferation, migration, and maintenance of multipotency.27,28 In particular, PDGFD functions as a potent angiogenic growth factor. 29 All PDGFs were upregulated in FEC and FEF compared to IZ in this study, with PDGFD transcript levels changing in a pattern that paralleled the functional assay results. Epithelial mitogen (EPGN) is a member of the epidermal growth factor (EGF) family and has been implicated in cell survival, proliferation, and migration. It has been shown to stimulate proliferation of epithelial cells.30,31 FGF2 and FGF5 belong to the fibroblast growth factor (FGF) family of secreted signaling proteins and function as key regulators of proliferation, differentiation, and survival in developmental, physiological, and pathological processes. 32 FGF2 is a potent angiogenic factor that stimulates proliferation in cultured murine cardiomyocytes and in aorta-derived vascular smooth muscle cells.33-36 FGF5 has been shown to facilitate proliferation in human tonsil–derived MSCs. 37 Hepatoma-derived growth factor (HDGF), originally identified as a mitogen from the human hepatoma cell line HuH-7, is a nuclear targeted protein, and has been demonstrated to have a growth-stimulating activity on fibroblasts, hepatoma cells, vascular smooth muscle cells, and endothelial cells.38-41 Insulin-like growth factor 1 (IGF1) is a potent mitogen for a multitude of cells including neural progenitor-like cells (NPCs), and hair follicle cells and acts as a regulator of bone growth and development.42-44 A key anabolic growth factor for articular cartilage, IGF-1 is expressed in growth plate chondrocytes45,46 and has been implicated in inducing chondrogenesis in in vitro studies47,48 and cartilage defects in vivo.49-51 C-fos-induced growth factor (FIGF) is a secreted factor related to the platelet-derived growth factor/vascular endothelial growth factor (PDGF/VEGF) family which has mitogenic and morphogenic effects on fibroblasts. 52 Placental growth factor (PGF) is a member of the VEGF subfamily and functions primarily as a regulator of angiogenesis. It has also been shown to stimulate proliferation in human epithelial cells.53,54 Amphiregulin (AREG) is also an EGF family member and has a mitogenic effect on several cell types including epithelial cells, fibroblasts, and immune cells.55-57 EGF containing fibulin like extracellular matrix protein 1 (EFEMP1) has been demonstrated to promote angiogenesis and proliferation of tumor cells.58,59 The upregulation of these growth factors in a manner concordant with that observed in the functional assays did not always reach statistical significance, but suggests that these could be potential candidates as regulators of cellular paracrine interactions between interzone and its environment.
The genes targeted in this study were selected and filtered based on established functional annotation related to cell proliferation. Transcriptome profiling broadly will likely identify additional genes of interest for the paracrine interaction between interzone cells and their skeletal environment, which can then be investigated in functional assays.
Supplemental Material
Fig_S1_600 – Supplemental material for Effect of Skeletal Paracrine Signals on the Proliferation of Interzone Cells
Supplemental material, Fig_S1_600 for Effect of Skeletal Paracrine Signals on the Proliferation of Interzone Cells by Parvathy Thampi, Rashmi Dubey, Rachael Lowney, Emma N. Adam, Sarah Janse, Constance L. Wood and James N. MacLeod in CARTILAGE
Supplemental Material
Fig_S2_600 – Supplemental material for Effect of Skeletal Paracrine Signals on the Proliferation of Interzone Cells
Supplemental material, Fig_S2_600 for Effect of Skeletal Paracrine Signals on the Proliferation of Interzone Cells by Parvathy Thampi, Rashmi Dubey, Rachael Lowney, Emma N. Adam, Sarah Janse, Constance L. Wood and James N. MacLeod in CARTILAGE
Supplemental Material
Fig_S3_600 – Supplemental material for Effect of Skeletal Paracrine Signals on the Proliferation of Interzone Cells
Supplemental material, Fig_S3_600 for Effect of Skeletal Paracrine Signals on the Proliferation of Interzone Cells by Parvathy Thampi, Rashmi Dubey, Rachael Lowney, Emma N. Adam, Sarah Janse, Constance L. Wood and James N. MacLeod in CARTILAGE
Supplemental Material
Supplementary_tables – Supplemental material for Effect of Skeletal Paracrine Signals on the Proliferation of Interzone Cells
Supplemental material, Supplementary_tables for Effect of Skeletal Paracrine Signals on the Proliferation of Interzone Cells by Parvathy Thampi, Rashmi Dubey, Rachael Lowney, Emma N. Adam, Sarah Janse, Constance L. Wood and James N. MacLeod in CARTILAGE
Footnotes
Supplemental Material
Acknowledgments and Funding
The authors wish to gratefully acknowledge the technical assistance of Dr. Tyler Kirby with the cell proliferation assay. We thank Dr. Barry Ball, Dr. Alejandro Esteller-Vico, Chad Tucker, and Kevin Gallagher for assistance with the collection of experimental equine samples. The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This research was supported financially by the Morris Animal Foundation, the Lourie Foundation, and the Geoffrey C. Hughes Foundation.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Procedures were performed under the approval of the University of Kentucky Institutional Animal Care and Use Committee (IACUC #2014-1215).
Animal Welfare
The present study followed international, national, and/or institutional guidelines for humane animal treatment and complied with relevant legislation.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.