
Abstract
Objective
Inflammation is a major player in the joint destruction process. Macrolide antibiotics have recently been found to have a novel anti-inflammatory function, but their effects on the joint are unknown. Our objective was to investigate the effect of macrolide antibiotic erythromycin on cartilage gene expression under inflammatory conditions as well as on joint pathology in an in vivo inflammatory joint destruction model.
Design
In our in vivo studies, mouse knee joints were injected with monosodium iodoacetate (MIA), a chemical that inhibits glycolysis and causes joint inflammation and matrix loss. Erythromycin was administered by daily intraperitoneal injection. Changes in joint cartilage and synovium were evaluated by histological analysis. In our in vitro studies, primary bovine articular chondrocytes were treated with erythromycin in the presence of pro-inflammatory cytokine IL-1β or lipopolysaccharide (LPS), and cartilage gene expression analysis was performed.
Results
Regional differences in cartilage matrix destruction along the medial-lateral axis were observed in joints of MIA-injected mice. Erythromycin treatment inhibited cartilage matrix loss and synovitis in these joints. In addition, erythromycin inhibited IL-1β and LPS-induced expression of MMPs and iNOS, as well as the positive regulatory loop between IL-1β and Toll-like receptor 4 (TLR4) in articular chondrocytes. Furthermore, erythromycin prevented LPS-induced NF-κB activation, a key mediator of TLR4-mediated cartilage destruction process.
Conclusions
Erythromycin has the ability to inhibit catabolic gene expression mediated by IL-1β and TLR4 in chondrocytes in vitro and maintains cartilage matrix levels in experimental inflammatory joint destruction in vivo, suggesting that it possesses a chondroprotective activity.
Introduction
The joint is an important structure that supports the body and facilitates movement. However, it is also the target of arthritis, a disease that causes widespread disability. The most common form of arthritis is osteoarthritis (OA), which is characterized by cartilage matrix loss, synovitis, and other pathological changes of joint tissues.1-3 Yet, there are few treatment options for OA due to the limited understanding of OA development and heterogeneity in its pathology. 4 Local inflammation is widely considered to be associated with OA development and cartilage degradation.5-7 In fact, interleukin-1-beta (IL-1β) is a prominent pro-inflammatory cytokine that causes cartilage matrix loss. Antagonizing IL-1β directly in the joint leads to matrix protection in posttraumatic experimental OA.8-10 However, a recent report indicated that while IL-1β in human synovial fluid was up-regulated in the early phase of OA, its level was much lower in late OA. 11 Furthermore, systemic IL-1β inhibition in mice did not lead to a reduction in joint inflammation and cartilage destruction in experimental OA, or symptom relief in a human clinical trial, 12 suggesting that other catabolic factors in the joint may be promoting continued cartilage damage in OA. It is now established that Toll-like receptors (TLRs), especially TLR2 and TLR4, can be induced by IL-1β and are upregulated in OA.13-16 Among the TLRs, TLR4 seems to be especially important in joint maintenance, as local IL-1β-driven joint pathology was seen to be dependent on TLR4 activation rather than TLR2 activation. 17 Downstream of these inflammatory stimuli lies the nuclear factor-kappa B (NF-κB) signaling pathway, a key mediator whose activation leads to the induction of cartilage matrix degrading enzymes such as matrix metalloproteinases (MMPs) and aggrecanases, resulting in subsequent matrix degradation. 6
Recently, certain macrolide antibiotics such as erythromycin (EM), clarithryomycin, and roxithromycin were shown to have anti-inflammatory activities in chronic inflammatory conditions such as diffuse panbronchiolitis, rheumatoid arthritis and in debris-induced inflammatory osteoclastogenesis in bone.18-20 A derivative of erythromycin without antibiotic function still suppresses NF-κB activity in airway epithelial cells, 21 suggesting that its anti-inflammatory activity is independent of its antibiotic function. Among the macrolide antibiotics, only EM has been tested in a human OA clinical trial, which showed EM enhanced the effect of acetaminophen in reducing joint effusion and pain over the course of 4 months. 22 However, whether joint cartilage integrity was improved or whether EM alone has any effects on OA progression was not assessed in this study. Therefore, the role of EM on articular cartilage maintenance is still not known.
In this study, we utilized the monosodium iodoacetate (MIA)–induced murine inflammatory joint destruction model to assess the effect of EM on joint cartilage integrity in vivo. As a specific antagonist of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), MIA inhibits glycolysis and ATP production, which are changes that have been found in OA.23-26 Injecting MIA into the joint is known to result in cell death, joint cartilage matrix loss and synovitis.23-26 As MIA induces robust inflammation that subsequently subsides within days, it recapitulates the inflammatory aspect of joint destruction seen in patients after traumatic joint injury. 27 Consistently, it has been reported that administering MIA to chondrocytes resulted in increased MMP13 expression in vitro. 28 In this study, we evaluated the effect of EM treatment on cartilage matrix loss and synovitis in the MIA model in vivo and investigated the biochemical mechanisms by which EM acts in primary articular chondrocytes in vitro.
Materials and Methods
Experimental Animals
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committees at Tufts University. Wild-type CD1 male mice were purchased from Charles River Laboratories. Mice were caged in groups under the standard conditions with a 12-hour light/dark cycle and standard chow diet. 8 week old mice were anesthetized by isoflurane/O2 inhalation and received a single intra-articular injection of MIA (Sigma) using a 30G 0.5-inch needle with 62.5 μg MIA dissolved in 5 μL of sterile phosphate buffered saline (PBS). This concentration was because it was the lowest of the 3 concentrations tested (250, 125, and 62.5 μg) that elicited matrix loss (data not shown), which is much lower than the amount used for pain analysis (0.5-1 mg).29,30 EM (Sigma) was delivered into the mice by daily intraperitoneal injection at 400 μg/day, starting from 1 day before MIA injection through 10 days after MIA injection when mice were euthanized. This dosage of EM was chosen based on the amount of EM commonly used to treat inflammation (e.g., in diffuse panbronchiolititis) in humans (400-600 mg/day), the body weight ratio of human and mouse (around 2000:1) and the half-life of EM (39 minutes in the mouse, 3.4 hours in the human).29,30 PBS-injected contralateral knees served as a negative control. A total of 20 mice were used in all experiments, including those injected with MIA or PBS for histology and mice used for gene expression analysis. Five knee joints per group were used.
Histological Analysis of Mouse Knee Sections
Mice were sacrificed 10 days after MIA injection, and their knee joints were fixed with 4% paraformaldehyde overnight, decalcified with 0.33 M ethylenediaminetetraacetic acid and paraffin embedded. The medial half of the knee joint was sectioned sagittally at 5-μm thickness. Sections were stained with 0.1% Safranin O and counterstained with hematoxylin and fast green. Percentage matrix loss was quantified by calculating the surface areas that exhibit Safranin O loss over total surface area, as delineated by ImageJ. OARSI (Osteoarthritis Research Society International) scores were determined based on the percentage loss of Safranin O staining using the established OARSI scoring system 31,32. Cellularity was evaluated by the DAPI (4′,6-diamidino-2-phenylindole) staining in comparison with bright field images. A loss of DAPI staining in the lacuna (as shown in the bright field) indicated cell loss. The severity of synovitis was evaluated according to the commonly used scoring system 33,34, which has the following three criteria: enlargement of the synovial cell layer (0-3 points), cellularity of resident cells (0-3 points) and inflammatory infiltration (0-3 points). When the points are added up, a full score (9 points) indicates severe synovitis and a lower score indicates less synovitis. All scorings were performed blinded.
Primary Bovine Articular Chondrocytes In Vitro Culture
Primary bovine articular chondrocytes (BACs) were isolated from the articular surface of the fetlock joints as previously described. 35 Briefly, cartilage pieces were digested with 1 mg/mL bovine hyaluronidase (Sigma) for 15 minutes followed by 0.25% trypsin (Sigma) digestion for 30 minutes and 2 mg/mL collagenase (Sigma) digestion for 15 hours. Single cell suspension was obtained by passing the cells through a 40 μm cell strainer (BD Biosciences). Cells were cultured in DMEM supplemented with 10% fetal bovine serum (Hyclone) and antibiotic-antimycotic (Invitrogen) and used at passage 1 for the study. For in vitro studies, chondrocytes were cultured on tissue culture dishes, and were pretreated with EM (Sigma; 0.2 or 1 μg/mL) for 3 hours prior to treatments of 1 ng/mL IL-1β (Peprotech) or 100 ng/mL lipopolysaccharide (LPS; Sigma) for 4 days.
Cytotoxicity Assay of Erythromycin
Chondrocytes were seeded on tissue culture dishes and treated with EM (0.2, 1, 10, and 100 μg/mL) for 4 days before being subjected to Live/Dead Cell Viability/Cytotoxicity assay (Invitrogen) according to the manufacturer’s instruction. The ratios of dead cells/live cells were then determined. Data are reported as mean with 95% confidence intervals.
RT-qPCR Analysis
For experiments involving cultured primary chondrocytes, cells were grown for 4 days prior to RNA isolation using the RNeasy mini kit (Qiagen). For mouse articular cartilage isolation, the tibial plateau and femoral chondyle cartilage were isolated under the dissection microscope at 7 days post-MIA injection, and were cut into small pieces, and subsequently dissolved in the lysis buffer that accompanies the RNeasy mini kit. There were three mice/group (MIA and PBS injection). RNA was extracted from each joint individually, and for each knee joint, 100 to 200 ng of total RNA was obtained. RNA was reverse transcribed using the M-MLV reverse transcriptase (Invitrogen). Quantitative polymerase chain reaction (qPCR) was carried out using iTaq universal SYBR Green supermix on the iQ5 Real Time PCR Detection System (BioRad). Relative PCR values were calculated based on the values of the gene of interest and the reference gene using associated software. All primers were first tested for efficiency, and only melt curves resulting in single peaks with adequate and linear amplification were selected for subsequent chondrocyte gene expression analysis. Primer sequences are available on request. At least three independent experiments were performed in triplicate.
Western Blot Analysis
Cells were pretreated with erythromycin at 1 μg/mL for 3 hours followed by treatment of 100 ng/mL LPS for 30 minutes, 1 hour or 2 hours. Total protein was isolated using the standard RIPA buffer. Nuclear and cytoplasmic lysates were isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce) according to the manufacturer’s instruction. Protein concentrations were determined using the DC Protein Assay (Bio-Rad). Total protein (10 μg), cytoplasmic protein (10 μg) and nuclear protein (10 μg) were loaded. Primary and secondary antibodies are: rabbit anti-NF-κB/p65 (E498), mouse anti-IκBα (L35A5) (Cell signaling), mouse anti-β-actin (Abcam) and goat anti-rabbit or mouse IgG, (H + L) HRP conjugate (Millipore/Chemicon). Western Blot images were taken by EPSON Perfection scanner V500, and band intensity was quantified by ImageJ. At least 3 independent experiments were performed.
Immunocytochemistry Analysis
Cells were seeded at a density of 5 × 104 cells per well in 12-well plates and pretreated with EM at 1 μg/mL for 3 hours followed by 100 ng/mL LPS treatment for 1 hour. Cells were fixed with 4% paraformaldehyde and blocked with goat serum for 1 hour. Cells were incubated with rabbit anti-NF-κB/p65 (E498, Cell Signaling) antibody overnight at 4°C and 594 Alexa Fluor conjugated goat anti-rabbit antibody for 1 hour at room temperature. Nuclei were counterstained with DAPI.
NF-κB Transactivation Assay
Cells were seeded at a density of 105 cells per well in 12-well plates and transfected with 200 ng of a NF-κB element-driven luciferase reporter construct (generous gift from Dr. Gail Sonenshein, Tufts University) and 5 ng of Renilla luciferase plasmid (internal transfection control) using Fugene6 (Roche). 24 hours later, cells were treated with EM at 1 μg/mL for 3 hours followed by 100 ng/mL LPS treatment for 16 hours. Dual luciferase assay was conducted using the luciferase assay kit (Promega). NF-κB luciferase activity was normalized to Renilla luciferase activity. At least 3 independent experiments were performed in triplicate.
Microscopy Analysis
Bright-field and fluorescent images for histological and immunofluorescent analysis were taken with the Olympus IX71 inverted microscope and the Olympus DP70 digital camera.
Statistical Analysis
For parametric data, data are reported as mean with 95% confidence intervals and statistical analysis was performed using a paired Student’s t test or a 1-way analysis of variance followed by a post hoc Tukey test with P values <.05 considered significant. For nonparametric data (i.e., OARSI [Osteoarthritis Research Society International] and synovitis scoring), data are reported as a dot plot with median and statistical analysis was performed using the Kruskal-Wallis test followed by Mann-Whitney U test with Bonferroni correction. P values <.05 are considered significant. All statistical analyses were conducted using Prism 5 (GraphPad).
Results
Erythromycin Inhibits Cartilage Matrix Loss In Vivo
We hypothesized that EM has an anti-inflammatory activity that slows down joint destruction. To test this hypothesis in vivo, we used the MIA inflammatory joint destruction mouse model and delivered EM by daily intraperitoneal injection.25,36,37 Ten days after MIA injection, knee joints were evaluated by histological analysis. As different regions of the joint may be differentially affected in human OA,38,39 we divided the medial half of the knee joint into 3 zones along the medial-lateral axis ( Fig. 1A ).
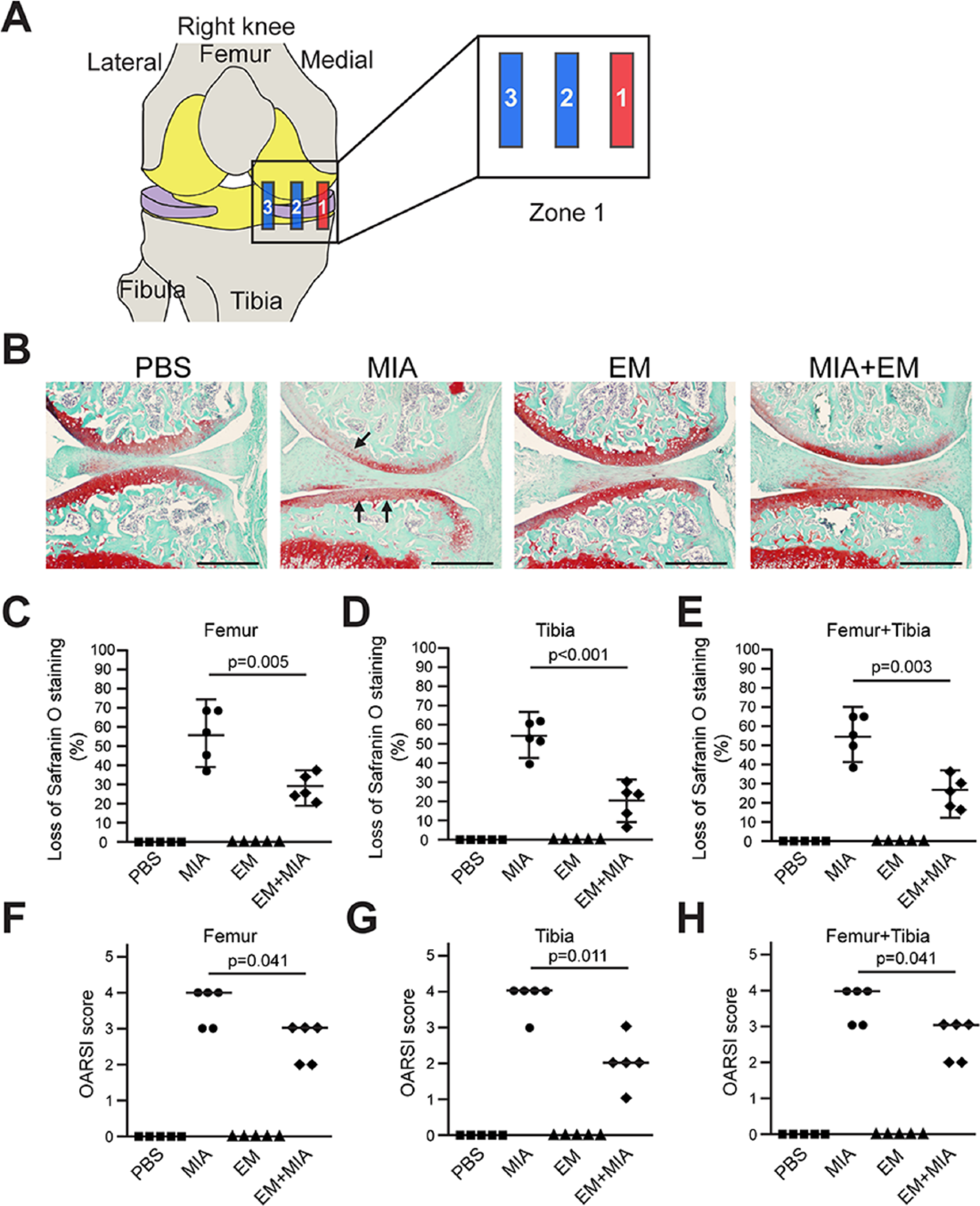
Erythromycin (EM) prevents monosodium iodoacetate (MIA)–induced articular cartilage destruction in the outer region of medial condyle of the mouse knee joint. (
In the most external region (zone 1), MIA treatment caused a significant loss of glycosaminoglycan, as evidenced by the loss of Safranin O staining ( Fig. 1B ). EM treatment in the control joint did not cause visible changes to cartilage matrix expression, but EM treatment in the MIA-injected joint largely diminished MIA-induced matrix loss ( Fig. 1B ). We quantified the extent of cartilage matrix loss using the established OARSI system, which categorizes OA severity into a scale of 1 to 6 based on percentage surface cartilage matrix loss. 31 We found that both the femur and the tibia showed significant articular cartilage matrix loss, while co-treatment with EM reduced such loss by half ( Fig. 1C-H ). In the middle region (zone 2), MIA-induced femoral cartilage damage was less severe compared to the external region (zone 1), but MIA-induced tibial cartilage damage was much more severe ( Fig. 2A-C ). EM co-treatment significantly reduced percentage cartilage matrix loss in this zone as well ( Fig. 2 ). However, when the percentage loss of surface cartilage was assigned an OARSI score, which rounds percentage loss into ordinal numbers (such as a loss of 26% to 49% to be given a score of 3), the differences between the treatment groups were reduced; as a result, the effect of EM on the femur was not statistically significant ( Fig. 2F ). EM inhibited cartilage matrix loss in the most internal region (zone 3) as well, although much less cartilage matrix is normally present as compared with other zones, and MIA-induced damage was also less severe ( Fig. 3 ).
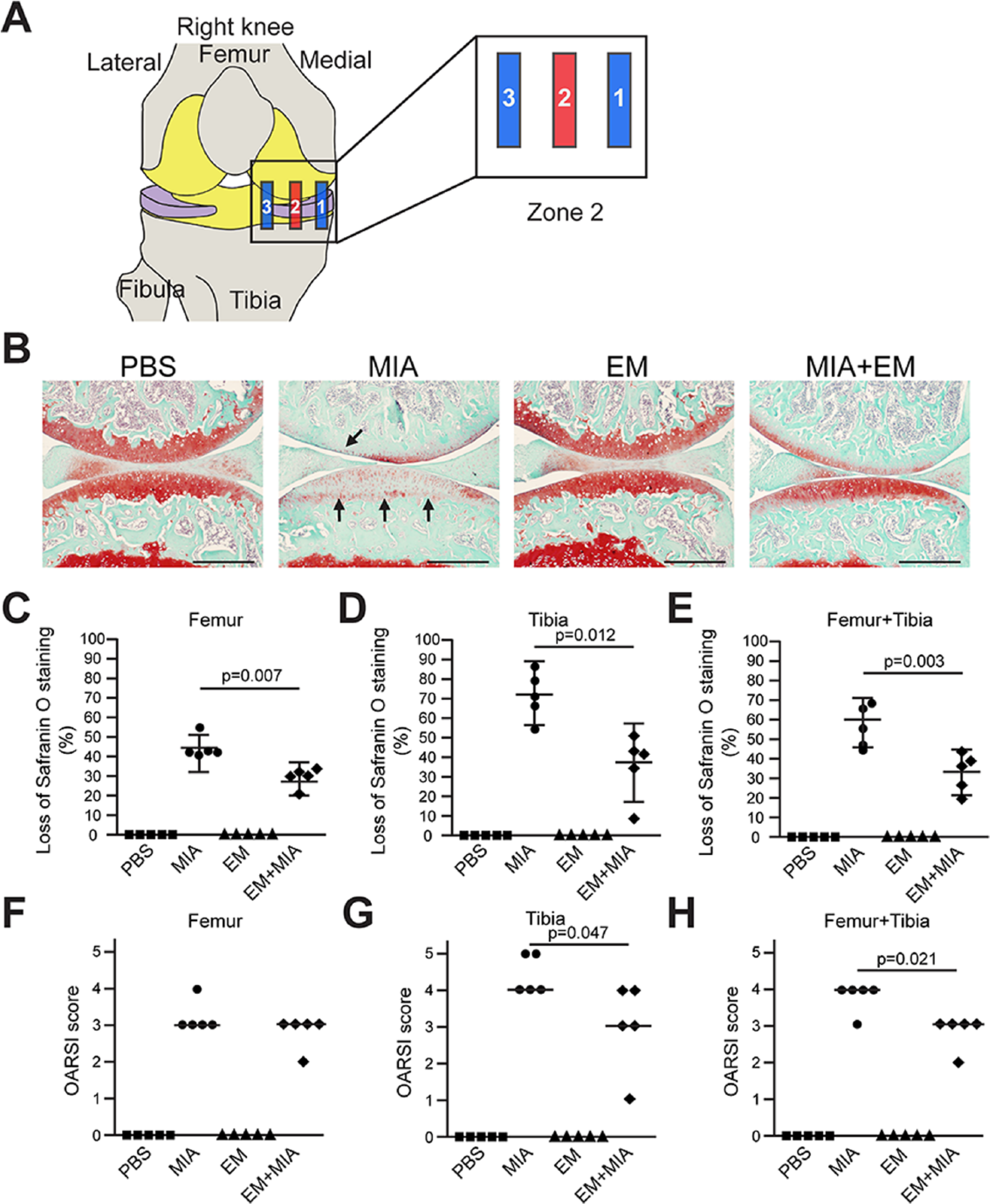
Erythromycin (EM) prevents monosodium iodoacetate (MIA)–induced articular cartilage destruction in the middle region of medial condyle of the mouse knee joint. (
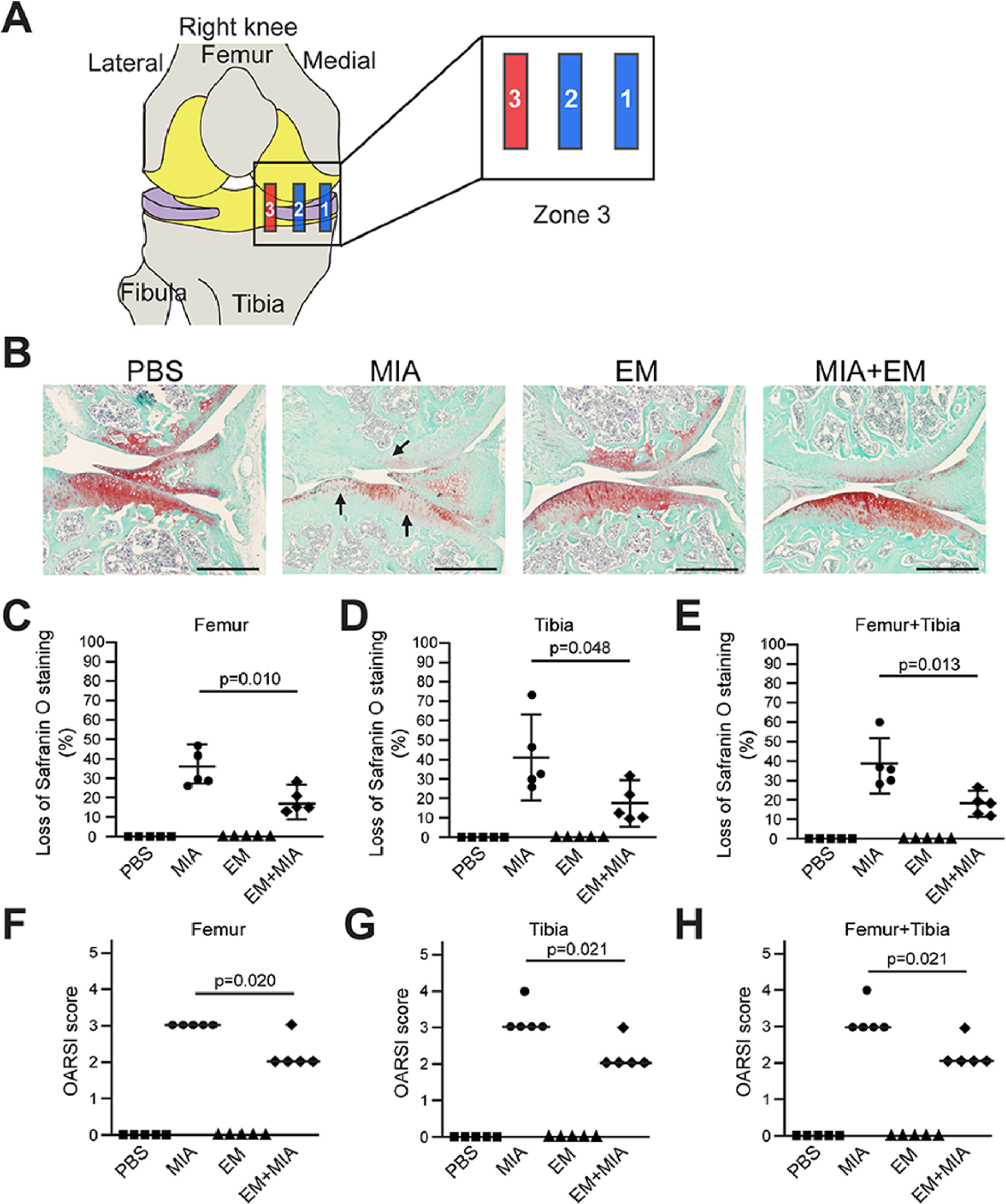
Erythromycin (EM) prevents monosodium iodoacetate (MIA)–induced articular cartilage destruction in the inner region of medial condyle of the mouse knee joint. (
Erythromycin Inhibits Cell Loss and Synovitis In Vivo
Chondrocyte cell loss is a characteristic change in OA. 40 Treatment with EM alone did not result in cell death, indicating minimum cell toxicity at this dosage. However, there was significant cell loss in MIA-injected knees (around 30%) ( Fig. 4A and B ). Significantly, treatment of EM reduced the percentage of cell loss to only 10% in MIA-injected knees, indicating that EM inhibits MIA-induced cell loss ( Fig. 4A and B ). In addition to cell loss, we also evaluated the synovium, since the synovium is an integral part of the joint and synovitis is also present in joint degeneration in OA.33,34 As shown in Figure 4C , the synovium of the PBS control knee exhibited a normal appearance consisting of 1 to 2 layers of cells, with a lower cellularity. However, the synovium in the MIA-injected knee clearly had more than 3 layers of cells with significantly increased cellularity, indicating synovium hyperplasia, even though clusters of inflammatory cells were rarely observed ( Fig. 4C and D ). EM injection alone did not result in any changes in the synovium ( Fig. 4C and D ). However, EM co-treatment with MIA substantially prevented synovium hyperplasia in the MIA knees ( Fig. 4C and D ). These results were further quantified in Figure 4D using the established synovitis quantification system.33,34
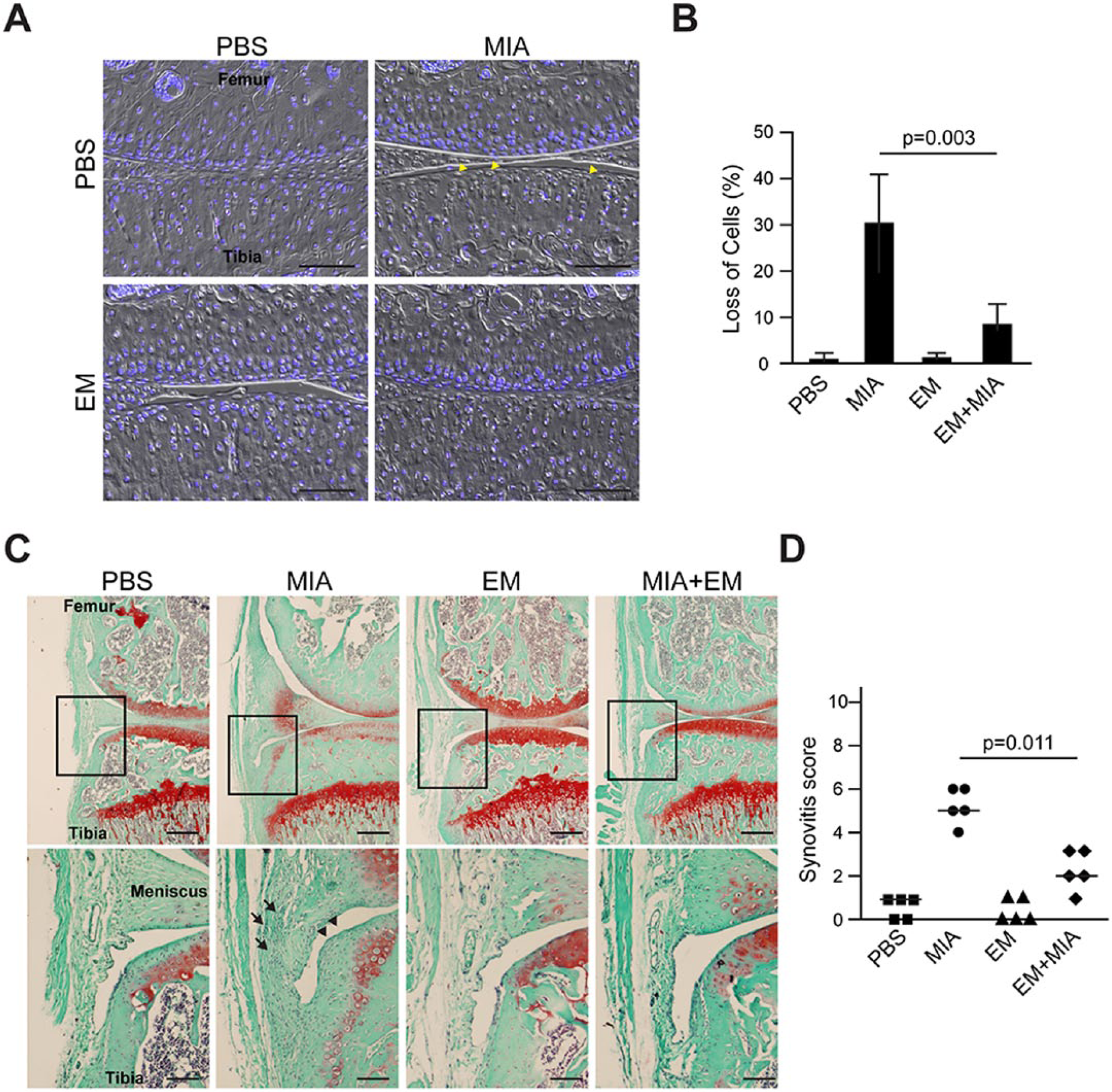
Histological analysis for cartilage cellularity and synovitis. (
Erythromycin Inhibits IL-1β- and TLR4-Mediated Catabolic Gene Induction in Chondrocytes
We next determined how EM may exert a protective effect on joint cartilage. RT-PCR analysis on articular cartilage isolated from the MIA-injected joints showed increased levels of pro-inflammatory cytokines IL-1β and TNFα as well as inflammatory mediator TLR4 ( Fig. 5A ). Since these pro-inflammatory stimuli are known to cause catabolic changes in chondrocytes, 6 we used them to perform in vitro experiments to test the effect of EM on chondrocytes. As very few primary adult murine articular chondrocytes could be obtained for culturing, primary bovine articular chondrocytes were used, which is a widely established in vitro culture system and allows large quantities of cells for qPCR and Western blot analysis.35,41 To determine the suitable dosage for EM treatment in vitro, we performed live/dead assays on chondrocytes treated with varying concentrations of EM to avoid potential complications of cell toxicity. As 0.2 and 1 μg/mL EM treatment resulted in minimal cell death, we proceeded to use these 2 dosages in future experiments ( Fig. 5B and C ). As expected, IL-1β inhibited the expression of cartilage matrix genes Col-II and Aggrecan, and induced MMP1, MMP13 and iNOS expression ( Fig. 5D-H ). EM pretreatment strongly inhibited IL-1β-induced expression of MMPs and iNOS, but had no effect on IL-1β-induced reduction of Col-II and Aggrecan ( Fig. 5D-H ). Because IL-1β has been shown to induce the expression of TLR4, 6 we also assessed TLR4 expression. As shown in Figure 5I , TLR4 was clearly induced by IL-1β, and this induction was abolished by co-treatment of EM.
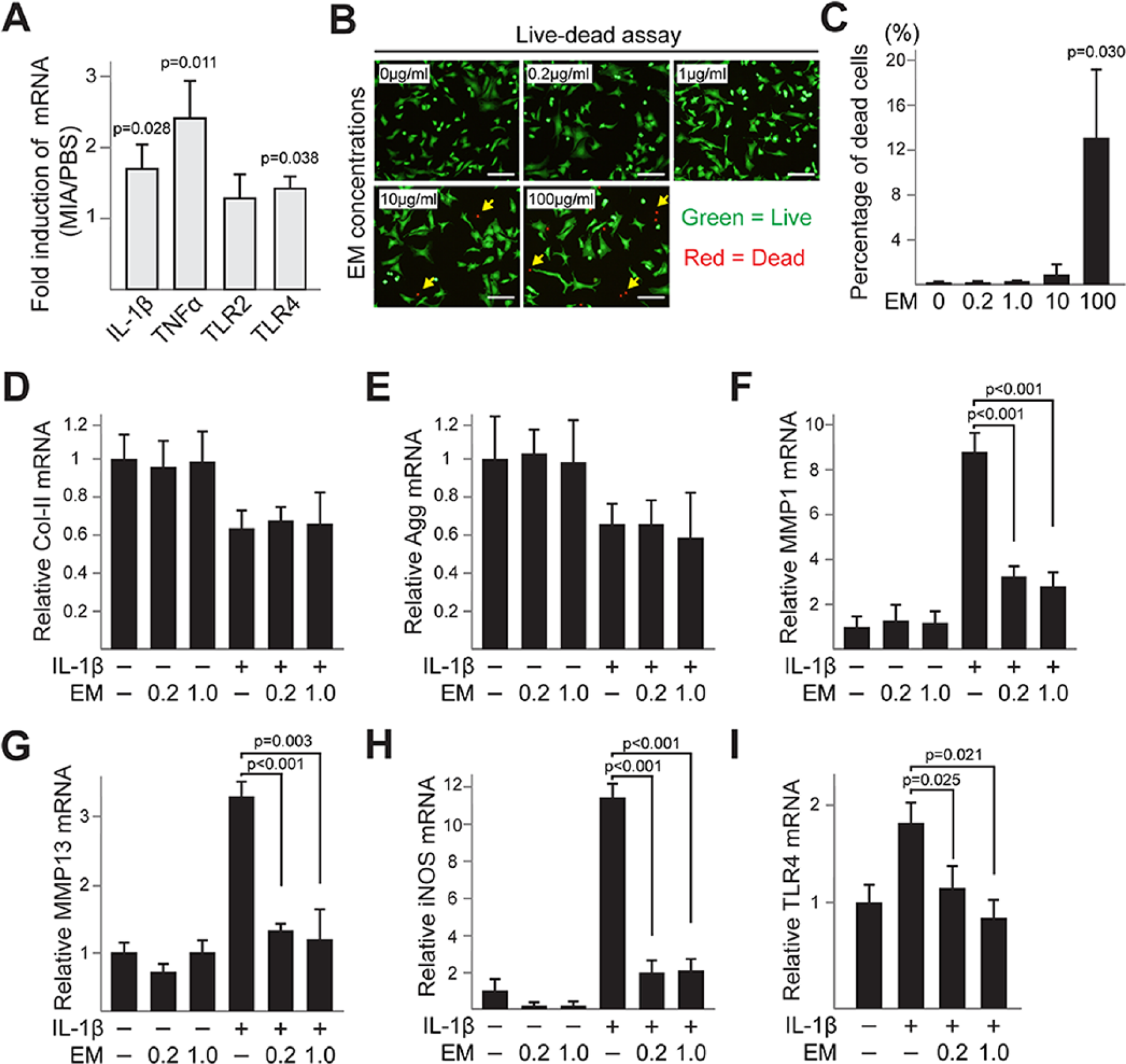
Erythromycin (EM) inhibits interleukin-1β (IL-1β)-induced catabolic gene expressions in bovine articular chondrocytes. (
To determine the effect of EM on TLR4-mediated catabolic gene expression, we utilized TLR4 receptor-specific agonist, LPS. 42 Since 1.0 μg/mL EM rather than 0.2 μg/mL showed a slightly stronger inhibitory effect on IL-1β activity in the previous experiments ( Fig. 5 ), 1.0 μg/mL EM was used for subsequent experiments. Similar to IL-1β, LPS inhibited the expression of cartilage matrix genes Col-II and Aggrecan, and strongly induced the expression of catabolic genes MMP1, MMP13 and iNOS ( Fig. 6A-F ). EM was also not able to inhibit LPS-mediated Col-II and Aggrecan induction ( Fig. 6A and B ), but significantly inhibited the expression of MMP1, MMP13 and iNOS induced by LPS ( Fig. 6C-E ). In addition, EM treatment reduced LPS-induced IL-1β expression, as well as IL-1β-induced TLR4 expression, suggesting that EM inhibits a positive regulatory loop between IL-1β and TLR4 ( Fig. 6F and G ).
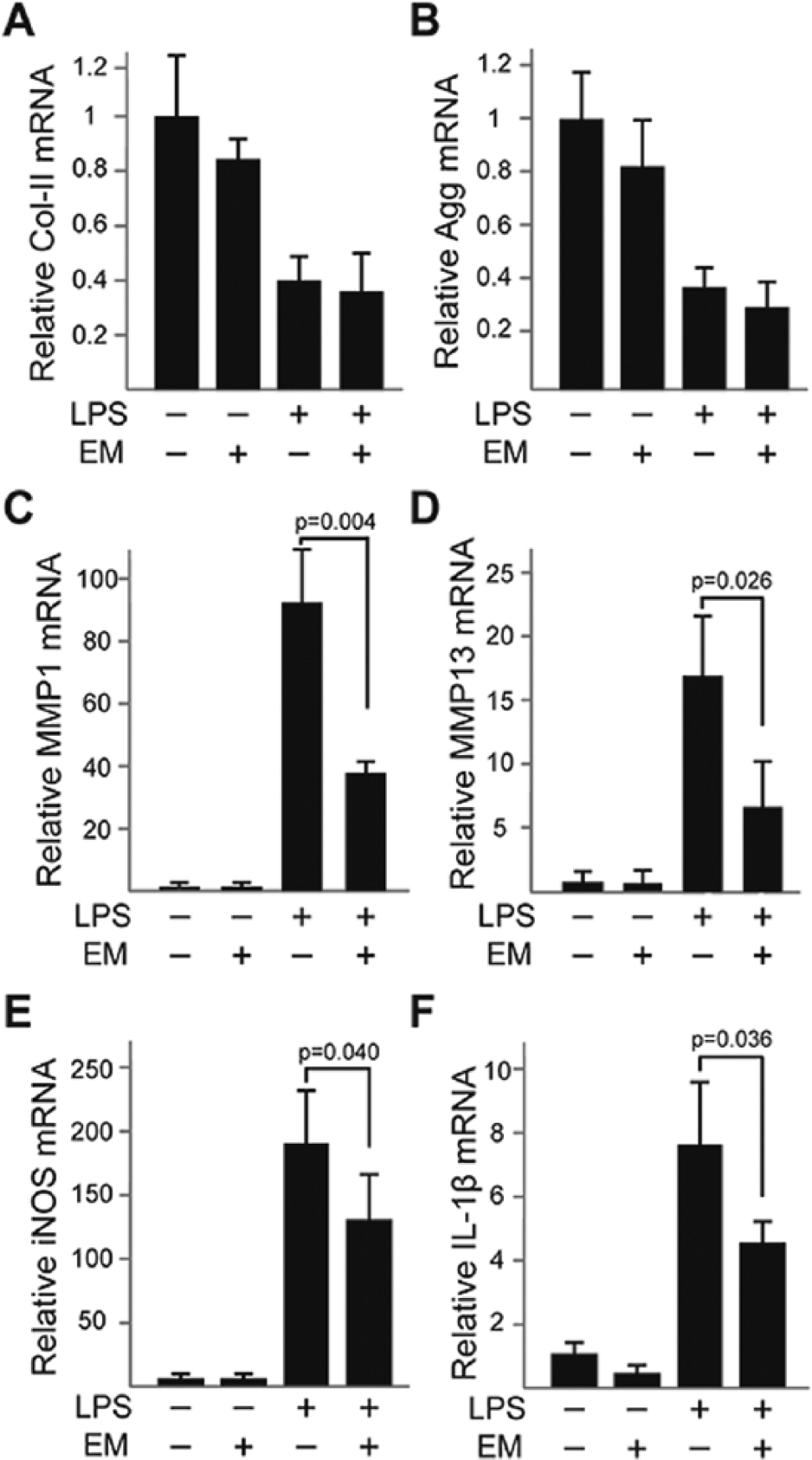
Erythromycin (EM) inhibits Toll-like receptor 4 (TLR4)–mediated catabolic gene expressions in bovine articular chondrocytes. (
To further determine the mechanism by which EM inhibits TLR4-mediated catabolic gene expression, we assessed the canonical NF-κB pathway, which mediates TLR4 signaling. 6 Typically, canonical NF-κB signaling activation involves IκBα degradation, resulting in subsequent translocation of NF-κB (subunits p65 and p50) into the nucleus. 6 To determine the timing of NF-κB activation in our system, we treated the chondrocytes with LPS for 30, 60, and 120 minutes, and assayed IκBα degradation by Western blot ( Fig. 7A and B ). The IκBα protein level was significantly reduced at 60 min and recovered to basal levels by 120 minutes ( Fig. 7A ). Consistently, a significantly increased NF-κB/p65 protein level in the nucleus was observed at 60 minutes ( Fig. 7B ). Therefore, we selected 60 minutes as the time frame for our subsequent studies. EM alone did not affect IκBα and p65 protein levels ( Fig. 7C ), but diminished LPS-induced p65 nuclear localization without affecting IκBα degradation ( Fig. 7C ). The effect on NF-κB nuclear localization was confirmed by immunocytochemistry, which showed reduced nuclear p65 localization in cells co-treated with EM and LPS, as compared with LPS alone ( Fig. 7D ). These observations were further confirmed using an NF-κB reporter construct, which showed much reduced luciferase activity when cells were co-treated with EM and LPS ( Fig. 7E ). The effects of EM on IL-1β and LPS-induced catabolic gene expressions in chondrocytes are shown in Figure 8 .
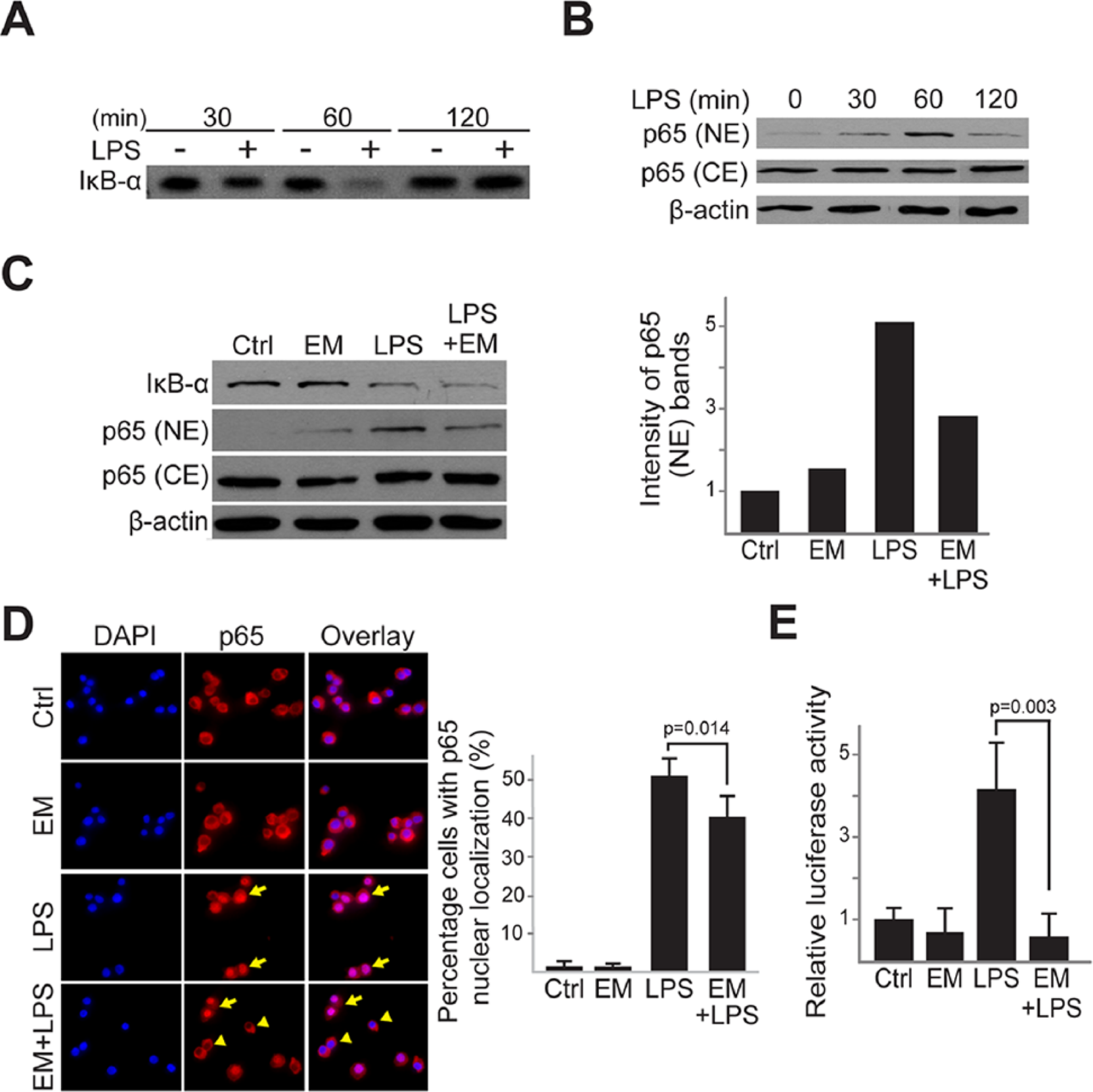
Erythromycin (EM) inhibits Toll-like receptor 4 (TLR4)–mediated NF-κB/p65 activation in bovine articular chondrocytes. (
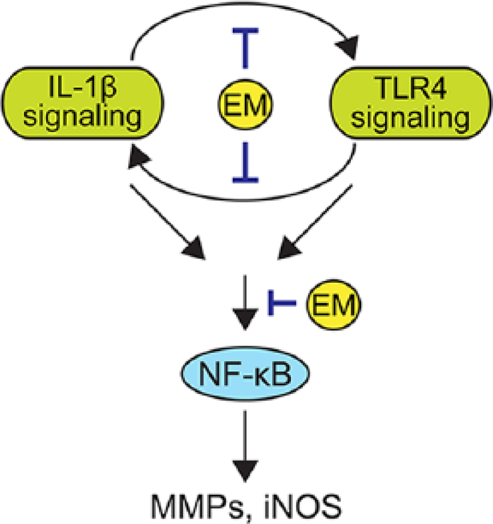
Schematic diagram of the effects of erythromycin (EM) on interleukin-1β (IL-1β) and lipopolysaccharide (LPS)–induced catabolic gene expressions in chondrocytes. IL-1β signaling and Toll like receptor 4 (TLR4) signaling form a positive regulatory loop activate the NF-κB pathway and the expression of catabolic gene such as matrix metalloproteinases (MMPs) or inducible nitric oxide synthase (iNOS). EM inhibits the nucleus translocation of NF-κB/p65 protein, and MMPs and iNOS expression as well as the positive loop between IL-1β and TLR4.
Discussion
Prior to this study, the tetracycline class of antibiotics (including doxycycline and minocycline) has been investigated as potential OA therapeutics. These antibiotics have the ability to chelate zinc at the active site of metalloproteinases, and were shown to reduce the levels of collagenase activity in rheumatoid arthritic (RA) joints. 43 In OA, doxycycline and a chemically modified tetracycline (CMT-7) were found to reduce cartilage loss in vivo in the dog and the guinea pig, respectively.44,45 Moreover, doxycycline treatment seemed to reduce joint space narrowing in human OA trials; however, in these trials, symptom relief such as pain and swelling was minimal.46,47 As a result, tetracycline-like antibiotics were not pursued further for treating OA.
In this study, we show that EM can inhibit catabolic gene expression in primary chondrocytes as well as joint degeneration in vivo. This is the first report of any macrolide antibiotic on maintaining cartilage stability in experimental OA. While OA in the MIA model is artificially induced, this model exhibits the typical inflammatory aspects of traumatic injury–induced joint destruction, including cell death, matrix loss and synovitis. As there is a robust early inflammatory phase that eventually subsides, this model is not considered a systemic rheumatoid arthritis model, but rather a localized inflammatory joint destruction model.36,37,48 Indeed, we have found increased mRNA levels of IL-1β, TNFα and TLR4 in the articular cartilage after 7 days of MIA injection. However, the extent of their induction was modest, which may be related to the relatively late timing of tissue harvest. Another feature of human joint destruction in OA is the regional differences of cartilage loss within the joint.38,39 Yet, this feature was rarely discussed in reported studies using animal OA models.
We carried out a thorough study analyzing the medial condyle of the femur and the medial plateau of the tibia in areas along the medial-lateral axis. In our normal knee joint samples, there was less Safranin O staining in the innermost region (zone 3), which is least covered by the meniscus. Consistent with this observation, Little et al. 49 also reported less matrix staining in this zone. MIA injection caused much more tibial cartilage damage in the middle region (zone 2) and the least in the inner region (zone 3). We also observed more femoral cartilage damage in the outer region than the inner zones. These are also consistent with the magnetic resonance imaging analysis of human OA cartilage,38,39 supporting the validity of this animal model. Such topographical differences could result from differential mechanical stress and the different proteoglycan synthesis of the chondrocytes in these regions.50-52 Remarkably, EM inhibited cartilage loss in all these regions, which is conceivable as EM is delivered systemically through intrapertoneal injections. Since EM was introduced into the mice one day before MIA treatment, it is not clear whether EM inhibits the initiation or the progression of joint destruction. It will be interesting to apply EM after MIA treatment to distinguish these possibilities. A limitation of this study is that the MIA inflammatory model normally develops joint destruction at a much faster pace, and that we have used very young mice and have not followed a longer time course in this inflammatory joint destruction model. As OA develops over a long period of time and is more commonly found in older individuals, it is important to perform follow-up experiments using older mice and evaluate them at later time points. While MIA mimics the inflammatory aspects of injury-induced OA, it does not involve a direct physical injury to the knee joint. Thus, it will be important to follow up with surgery-induced OA models (such as destabilization of the medial meniscus, and anterior cruciate ligament transection). Furthermore, it will be interesting to compare the effect of EM with other antibiotics within the macrolide class of antibiotics or those in the tetracycline class.
In our in vitro studies, we showed that EM has the ability to inhibit catabolic activities in chondrocytes. EM inhibited the positive regulatory loop between IL-1β and TLR4 both in terms of activity and gene expression. Although multiple studies showed that intra-articular injection of an IL-1β antagonist inhibited injury-induced OA,8,9 no results of OA manipulation on IL-1β knockout animals have been demonstrated. It was reported that the knockout of TLR2 and TLR4 mildly protect against meniscectomy-induced OA, 53 suggesting that TLRs may be required for OA development. However, the lack of a complete rescue in OA progression in the TLR KO indicates that other factors are also involved in this process. A limitation of our study is that we did not use natural TLR4 ligands in our study, although we did use a TLR4-specific agonist (i.e., LPS). 16 Among the natural ligands of TLR4 are low molecular weight hyaluronan and high mobility group box chromosomal protein 1 (HMGB1), although they are not necessarily specific to TLR4. 54 Therefore, future experiments would involve testing whether EM can inhibit TLR4 signaling induced by these natural ligands in chondrocytes. Our in vitro analysis also indicated that EM inhibits NF-κB activation, providing a plausible mechanism for its inhibitory effects on the activities of inflammatory stimuli. As NF-κB activation takes place as early as 30 minutes following IL-1β treatment, applying EM at the same time of IL-1β treatment might not allow sufficient time for EM to inhibit NF-κB nuclear localization. Thus, EM was applied before IL-1β administration. While EM prevented NF-κB/p65 nuclear localization and activity, it did not seem to affect IκBα levels. It is possible that since Western blot analysis is semiquantitative, subtle changes in IκBα proteins were not detected. It is also possible that EM regulates NF-κB nuclear localization without affecting IκBα. In other cell types, several compounds were found to inhibit p65 nuclear localization, but were not found to cause IκBα degradation.55,56 However, in all these cases, the underlying mechanisms are still not known. It is possible that EM affects nuclear shuttling of p65 and NF-κB regulators, 57 which would be our next step of investigation. Interestingly, EM did not inhibit NF-kB activity in synovial cells from rheumatoid arthritis patients, although it did inhibit Cox-2 expression, suggesting that EM may have different effects on different cell types in the joint. 58
The direct target of EM on chondrocytes has not been established. Previous research showed that EM acts as an agonist to the motilin receptor, a receptor involved in gastrointestinal movement. 59 However, motilin and its receptors are pseudogenes in rodents60,61; thus, the effect from our mouse experiments cannot possibly be due to its binding to the motilin receptor. Identifying the direct target of EM would be a key area in our future study. Despite these unanswered questions, in this study, we thoroughly analyzed the effect of EM on cartilage matrix loss and synovitis in an in vivo model of OA, providing insights into devising strategies for inhibiting cartilage loss and promoting cartilage repair. While prolonged systemic antibiotic use poses risks to both the individual patient as well as public health, such concerns may be addressed with EM delivery in a localized fashion or evaluating the efficacy of a non-antibiotic derivative of EM in OA. This derivative has already demonstrated anti-inflammatory activity in the lung, 21 and in following with current research here, may offer great potential as a therapeutic for halting OA progression.
Footnotes
Acknowledgments and Funding
We thank Dr. Gail Sonenshein (Tufts University) for providing us with the NF-κB luciferase reporter construct. We thank Daisy Nakamura, Averi Gibson, and Carrie Hui Mingalone (Tufts University) for critically reviewing this article. This work has been supported by grants to LZ from the National Institutes of Health (AR054611), the National Science Foundation (CBET-0966920), and a grant from the Tufts Clinical Translational Science Institute (UL1TR001064). The funding agencies had no role in the design, analysis or interpretation of the data or in the writing of the article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval for this study was obtained from the Institutional Animal Care and Use Committees at Tufts University (APPROVAL NUMBER/ID B2014-11).
Animal Welfare
The present study followed international, national, and/or institutional guidelines for humane animal treatment and complied with relevant legislation.