
Abstract
Ect2, a Rho guanine nucleotide exchange factor (RhoGEF), is atypical among RhoGEFs in its predominantly nuclear localization in interphase cells. One current model suggests that Ect2 mislocalization drives cellular transformation by promoting aberrant activation of cytoplasmic Rho family GTPase substrates. However, in ovarian cancers, where Ect2 is both amplified and overexpressed at the mRNA level, we observed that the protein is highly expressed and predominantly nuclear and that nuclear but not cytoplasmic Ect2 increases with advanced disease. Knockdown of Ect2 in ovarian cancer cell lines impaired their anchorage-independent growth without affecting their growth on plastic. Restoration of Ect2 expression rescued the anchorage-independent growth defect, but not if either the DH catalytic domain or the nuclear localization sequences of Ect2 were mutated. These results suggested a novel mechanism whereby Ect2 could drive transformation in ovarian cancer cells by acting as a RhoGEF specifically within the nucleus. Interestingly, Ect2 had an intrinsically distinct GTPase specificity profile in the nucleus versus the cytoplasm. Nuclear Ect2 bound preferentially to Rac1, while cytoplasmic Ect2 bound to RhoA but not Rac. Consistent with nuclear activation of endogenous Rac, Ect2 overexpression was sufficient to recruit Rac effectors to the nucleus, a process that required a functional Ect2 catalytic domain. Furthermore, expression of active nuclearly targeted Rac1 rescued the defect in transformed growth caused by Ect2 knockdown. Our work suggests a novel mechanism of Ect2-driven transformation, identifies subcellular localization as a regulator of GEF specificity, and implicates activation of nuclear Rac1 in cellular transformation.
Keywords
Introduction
Rho family GTPases are molecular switches that act as signaling nodes to integrate extracellular signals and propagate intracellular signals. They control many normal cellular processes, including actomyosin remodeling, cell polarity, gene expression, and cell cycle progression.1,2 The best-studied members of this family are RhoA, Rac1, and Cdc42. These Rho GTPases are frequently overexpressed or dysregulated in tumors.3-5 Aberrant Rho GTPase activity regulates transformation, invasion, metastasis, and angiogenesis.3-5
GDP-bound Rho proteins are in the off-state, whereas GTP-bound proteins are in the on-state, in which they bind to their effector targets and transmit downstream signals. Guanine nucleotide exchange factors (GEFs) catalyze release of guanine nucleotides from GTPases, thereby enhancing binding of the more abundant cellular GTP. 6 While some GEFs activate only specific GTPases, others are more promiscuous.
Ect2 (epithelial cell transforming sequence 2) is a Rho family GEF capable of activating RhoA, Rac1, and Cdc42 in vitro, 7 but in cellulo it is more selective, in a context-dependent manner.8-14 Atypically for RhoGEFs, Ect2 contains 2 nuclear localization signals (NLSs) and has a prominent nuclear localization in interphase cells. 7 In contrast, Rho proteins are found outside the nucleus. 5 It has been proposed that Ect2 is auto-inhibited and sequestered from Rho GTPases in the nucleus of normal interphase cells, but becomes mislocalized to the cytoplasm in tumor cells, where its auto-inhibition is lost, and where it then activates Rho family GTPases to drive transformation.8,15 However, the subcellular localization of Ect2/Rho GTPase interactions has never been directly investigated in tumor cells.
Here we used ovarian tumor cells to further examine the role of Ect2 in transformation. Aberrant Rho GTPase activity has been implicated in this tumor type.16-19 Ect2 is located on chromosome 3q26.1-26.2, 20 a common amplicon in ovarian tumors21,22; indeed, ovarian cancer has the second highest frequency of Ect2 amplification among human cancers to date. 23 Ect2 is also overexpressed at the mRNA level.22,24,25 However, the protein expression of Ect2 and its functional consequences have not been studied in ovarian tumors. We examined a patient tumor tissue microarray (TMA) and observed that Ect2 protein was strongly expressed predominantly in the nucleus of ovarian cancer cells. We have identified a requirement for Ect2 in ovarian cancer cell transformation, and a novel mechanism whereby Ect2 can activate Rac1 and can drive ovarian tumor cell transformation from within the nucleus.
Results
Nuclear localization of Ect2 correlates with advanced disease in human serous epithelial ovarian cancers
Protein expression and subcellular distribution of Ect2 in ovarian cancers has not been evaluated previously. We evaluated these properties in a previously validated ovarian TMA26-29 containing approximately 400 full-faced cores from ovarian tumors and non-matched normal ovarian cysts. We optimized the immunohistochemical protocol such that <5% of OVCAR8 cells with Ect2 knockdown stained positive for Ect2, using an Ect2 antibody that we had previously validated for specificity by immunoblot analysis (Suppl. Fig. S1A and S1B). Nuclear and cytoplasmic Ect2 expressions were scored independently for each core. Unexpectedly, we found that higher scores for nuclear expression correlated with more advanced serous epithelial tumors (P = 0.0001516; Fig. 1A), whereas those for cytoplasmic expression correlated with less advanced tumors (P = 0.0007163; Fig. 1B). Indeed, in serous cysts Ect2 was expressed at low levels in the cytoplasm but undetectable in the nucleus, whereas for most cells in advanced tumors, Ect2 was concentrated in nuclei (Fig. 1C). These results suggested that nuclear rather than cytoplasmic localization of Ect2 may be important for its oncogenic functions in this tumor type.
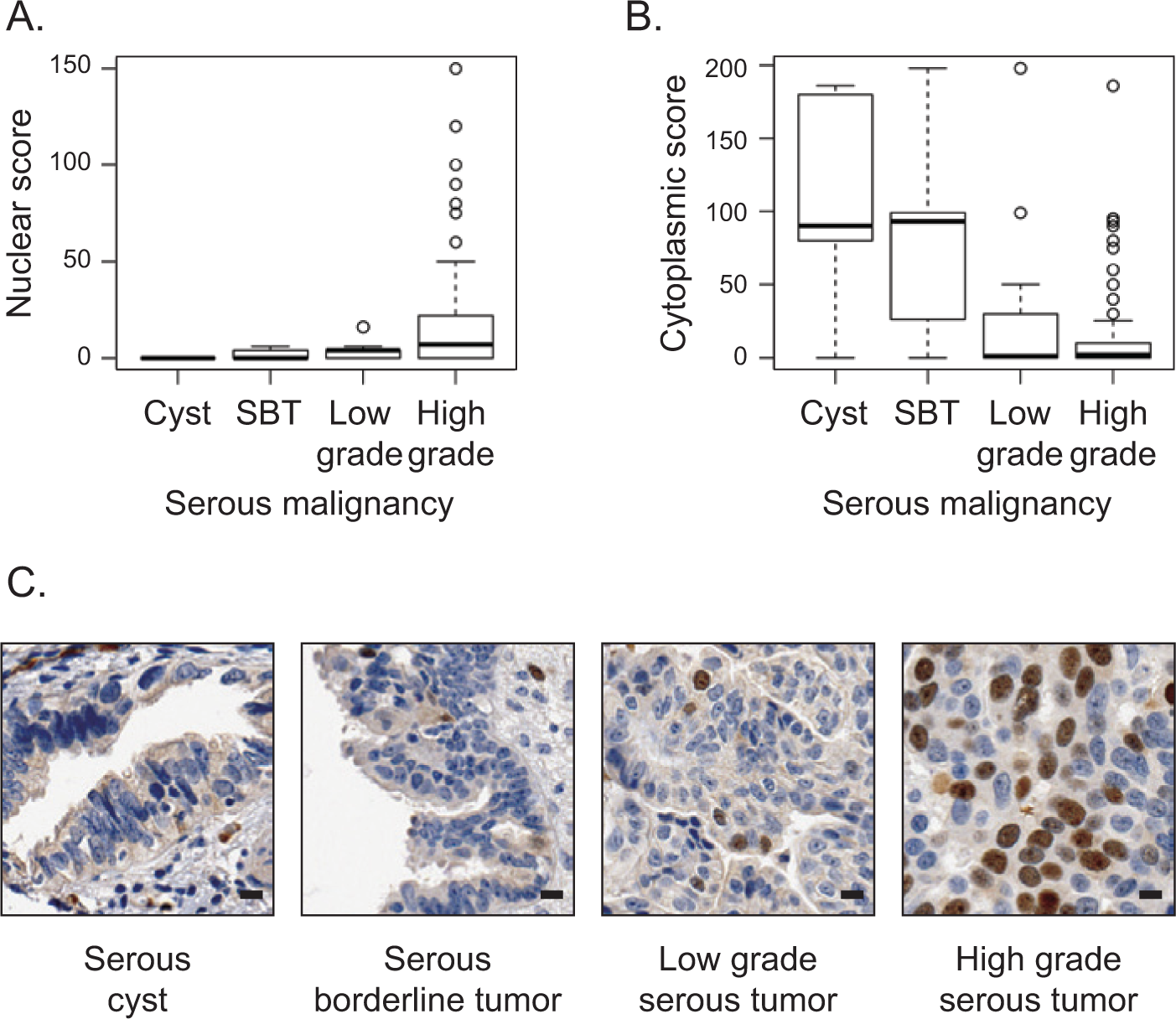
Nuclear localization of Ect2 correlates with advanced disease in human serous epithelial ovarian cancers. Ect2 subcellular localization was analyzed by immunohistochemistry (IHC) using a well-validated26-29 ovarian tissue microarray (TMA). Tumor cores were stained with Ect2 antibody (1:350; Millipore, Billerica, MA) and scored for percentage cells stained as well as intensity of staining in each location. Separate nuclear scores and cytoplasmic scores were calculated as described in Materials and Methods and binned according to disease severity. Quantification of Ect2 localization showed (
Ect2 is expressed predominantly in the nucleus of ovarian cancer cell lines
We turned to cell lines to study the localization and function of Ect2 in ovarian tumor cells directly. Ect2 was easily detected by immunoblotting in all 8 ovarian cancer cell lines tested (Fig. 2A). To assess its subcellular localization, we performed immunofluorescence, staining for endogenous Ect2, co-staining for DAPI as a nuclear marker, and imaging the cells using confocal microscopy. Specificity of the Ect2 antibody immunofluorescence signal was confirmed using Ect2 knockdown cells (Suppl. Fig. S1C). We observed a direct overlay of the vast majority of the Ect2 and DAPI signals in all cell lines examined (Fig. 2B and data not shown), indicating that Ect2 is predominantly nuclear, and supporting our observation from the TMA that nuclear Ect2 may be more relevant than cytoplasmic Ect2 for ovarian cancer cells.
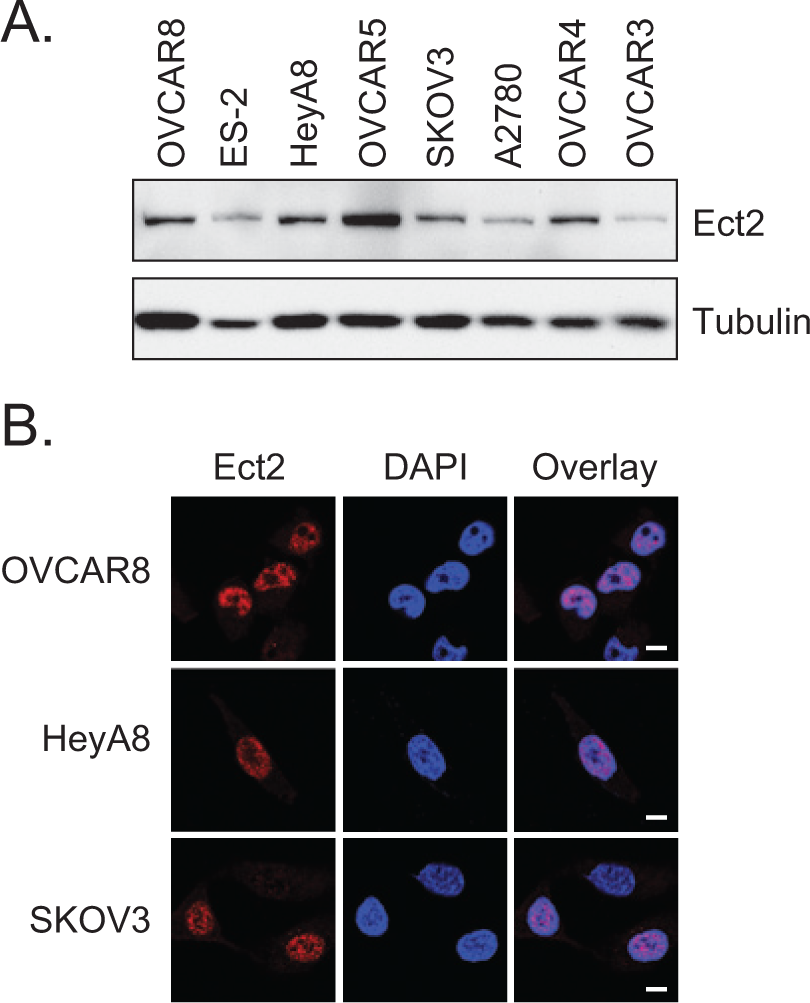
Ect2 is expressed predominantly in the nucleus of ovarian cancer cell lines. (
Ect2 is required for transformed growth of ovarian cancer cell lines
Anchorage-independent growth is a hallmark of the transformed phenotype and is classically measured by soft agar colony formation. 30 To determine the requirement for Ect2, we stably knocked down Ect2 using 2 independent shRNAs in cell lines that grow vigorously in an anchorage-independent manner. Both shRNAs robustly decreased expression of Ect2 in all cell lines, with Ect2 shRNA#3 showing more complete knockdown than shRNA#2 (98% and 84%, respectively, in OVCAR8 cells) (Fig. 3A). We then compared anchorage-independent growth of cells expressing non-targeted (NT) shRNA or Ect2 shRNA. Knockdown of Ect2 decreased soft agar colony formation of multiple cell lines, with the most striking effects seen in OVCAR8 cells (Fig. 3B, *P < 0.05). Thus, Ect2 is necessary for anchorage-independent growth of multiple ovarian cancer cell lines. We selected OVCAR8 for further study.
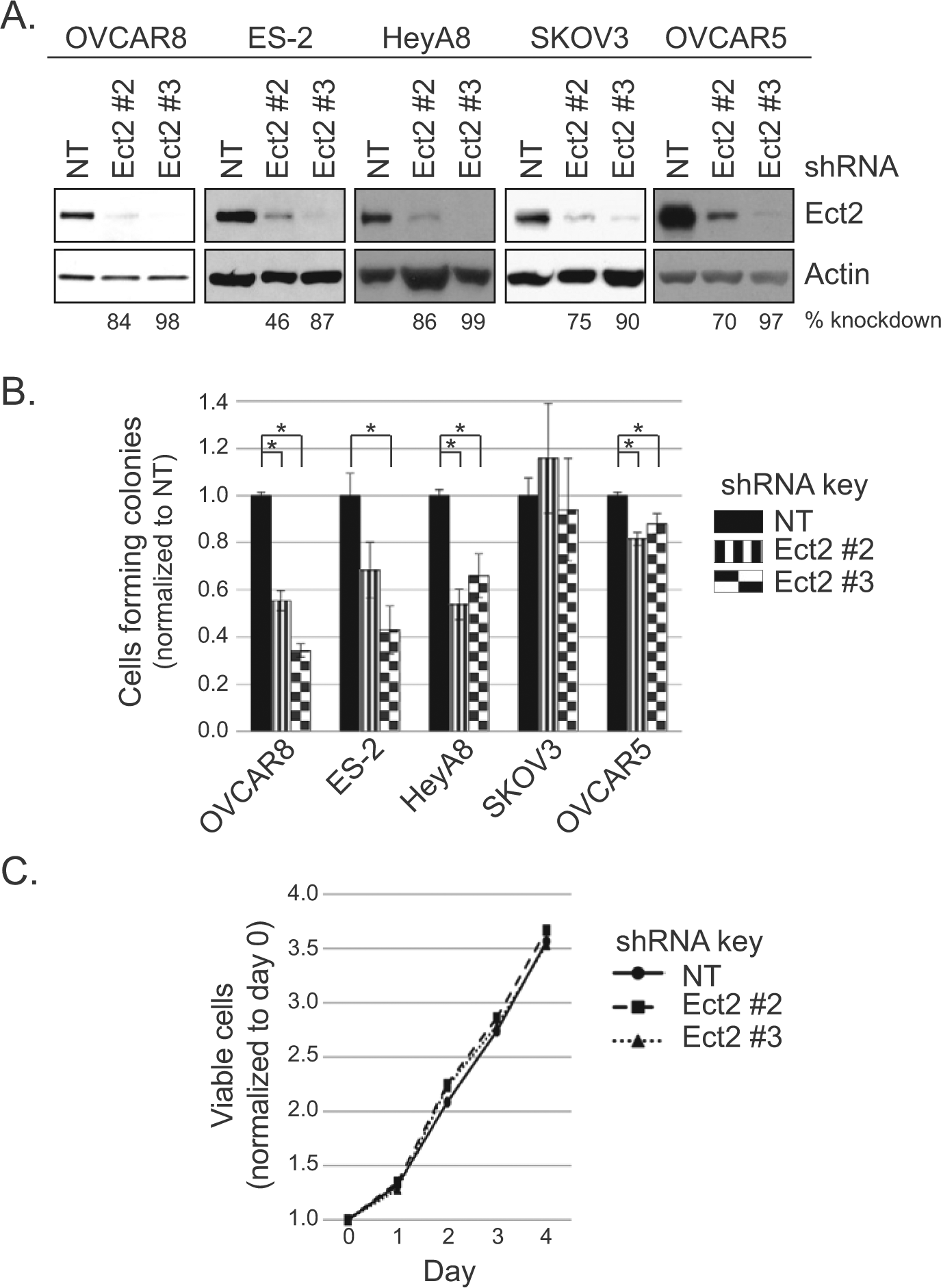
Ect2 is required for transformed growth of ovarian cancer cell lines. Our ovarian cancer cell lines that grow robustly in an anchorage-independent manner were evaluated for the importance of Ect2 in transformed growth. Cells were infected with lentiviruses expressing 1 of 2 independent shRNAs directed against Ect2, or non-targeting (NT) shRNA. Infected cells were selected in puromycin and colonies were pooled for further use. (
MTT proliferation assays revealed no significant differences between NT and Ect2 knockdown ovarian cancer cells (Fig. 3C). This indicates that the observed Ect2 dependence of anchorage-independent growth was not due simply to a general proliferation defect. And although Ect2 is known to play a critical role in cytokinesis of normal cells through activation of RhoA,10,31 its role in some tumor cells is less clear.11,15 Here, the level of multinucleated cells remained low upon knockdown of Ect2 (Suppl. Figs. S2 and S4), indicating that these ovarian tumor cells can still undergo cytokinesis efficiently when Ect2 is depleted.
RhoGEF activity is required for Ect2-mediated transformed growth
Ect2 is a RhoGEF with other functional domains 6 that may also play a role in its ability to drive transformed growth. To determine if the GEF activity itself is required, we first generated an shRNA-resistant, putatively GEF-deficient Ect2 mutant. Residues E428 and N608 in the catalytic Dbl homology (DH) domain of Ect2 are conserved among RhoGEFs, interact with the switch I and switch II regions of Rho GTPases, respectively, and are important for nucleotide exchange. 6 We mutated these residues to alanines (E428A, N608A). Both wild type (WT) and the DH-mutant Ect2 were tagged with the HA epitope so they could be monitored separately from endogenous Ect2. The localization of the DH-mutant was indistinguishable from WT (Suppl. Fig. S3). Using standard pulldown assays for active Rho GTPases, we then observed in 293T cells that these mutations decreased by 90% the ability of Ect2 to activate RhoA, and completely ablated Rac1 activation (Fig. 4A), validating that this Ect2 mutant is GEF-deficient on these substrates. Interestingly, these mutations did not prevent Ect2-induced activation of Cdc42, indicating that this activation is not dependent on these catalytic residues and is likely indirect.
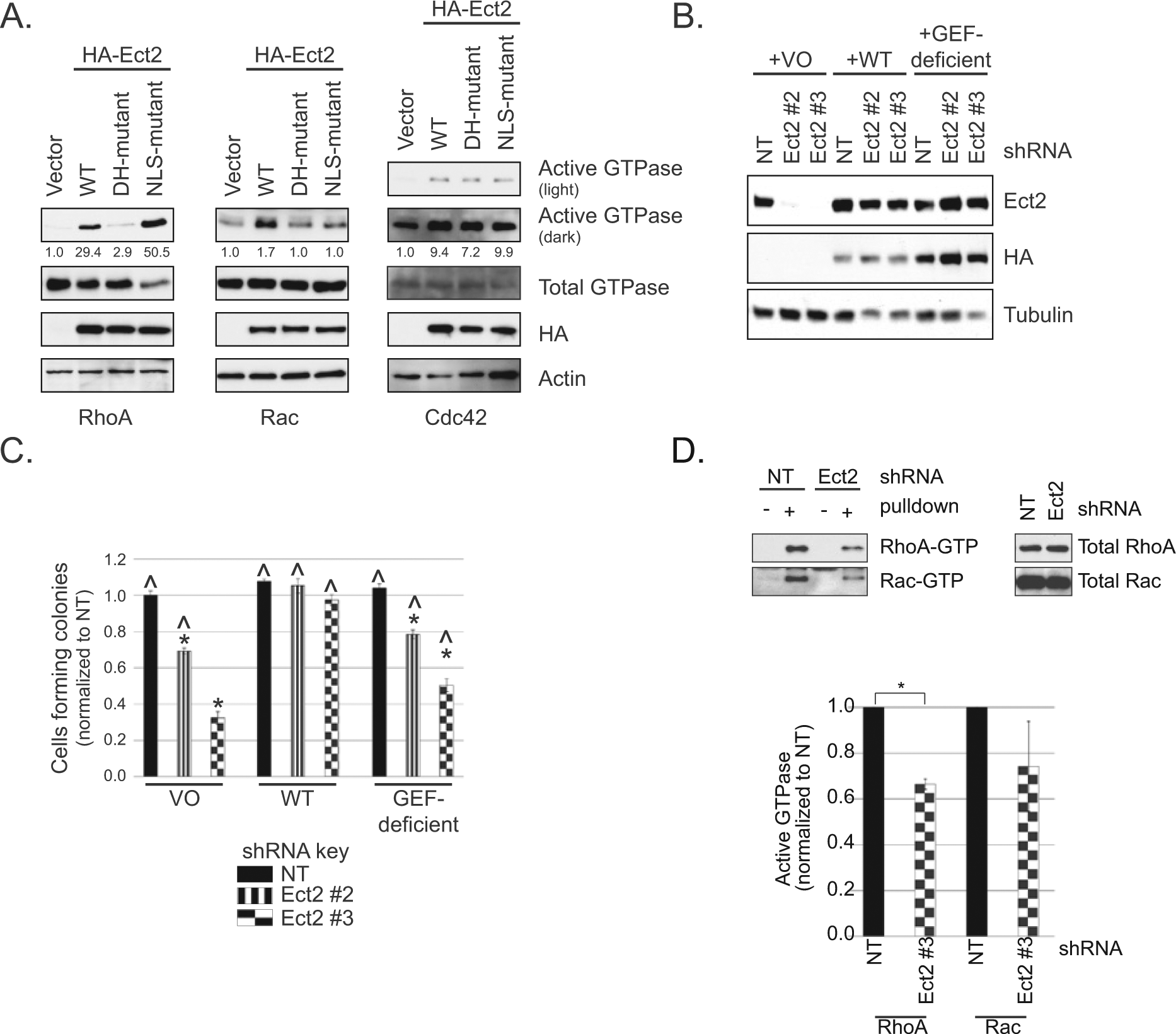
RhoGEF activity is required for Ect2-mediated transformed growth. (
We next assessed whether GEF-deficient Ect2 could rescue anchorage-independent growth. Ect2 expression was rescued in OVCAR8 Ect2 knockdown cells using either WT or GEF-deficient Ect2 (Fig. 4B). As expected, WT Ect2 rescued soft agar colony numbers to levels similar to those seen in cells without Ect2 knockdown, whereas the GEF-deficient mutant only partially rescued the phenotype (Fig. 4C). Thus, the RhoGEF activity of Ect2 is required to support full anchorage-independent growth in OVCAR8 cells.
We then sought to determine which Rho GTPases are activated by Ect2 in these cells. We compared the levels of active RhoA, Rac, and Cdc42 in whole cell lysates with or without Ect2 knockdown, using standard Rhotekin-RBD (for RhoA) and PAK-RBD (for Rac and Cdc42) pulldown assays. Upon Ect2 knockdown, RhoA activity decreased by about a third (*P < 0.05, Fig. 4D). We also saw a clear but more variable decrease in active Rac, despite the continued presence of multiple other RhoGEFs capable of activating these GTPases. Cdc42 activity remained constant (data not shown), consistent with the lack of effect of Ect2 GEF deficiency on Cdc42-GTP levels. These results indicate that Ect2 GEF activity is required for full RhoA and Rac activation in OVCAR8 cells at steady state and that other GEFs cannot fully compensate for loss of Ect2.
Predominantly nuclear localization is required for Ect2-mediated transformed growth
In ovarian cancer cells, Ect2 is a predominantly nuclear RhoGEF. Although RhoA and Rac1 are predominantly cytosolic proteins, 5 both have been observed to lesser extents in the nucleus.32-36 Loss of Ect2 GEF activity impaired ovarian cancer cell transformed growth and also impaired full RhoA and Rac activation. Collectively, these findings suggest that Ect2 could activate nuclear rather than cytoplasmic pools of RhoA and Rac to drive transformation. If so, depleting Ect2 selectively from the nucleus should impair anchorage-independent growth.
To address this possibility, we mutated the arginines in both NLSs of the shRNA-resistant Ect2 to alanines (R348,349,350,370,372A). We first evaluated subcellular localization using immunofluorescence and confocal microscopy. Co-staining for the HA-epitope tag and DAPI confirmed that the NLS-mutant had a greatly decreased nuclear localization and greatly enriched cytoplasmic localization compared to WT Ect2 (Fig. 5A). To quantitate the subcellular distribution of NLS-mutant Ect2, we then fractionated the cells and performed immunoblotting analyses. The results (Fig. 5B) demonstrated that only about 10% of the NLS-mutant was still localized to the nuclear fraction. The NLS-mutant was well-expressed and well-tolerated, such that cell lines stably expressing NLS-mutant Ect2 were easily generated (Suppl. Fig. S4A), without apparent effects on cell cycle distribution (Suppl. Fig. S4D) or multinucleation (Suppl. Fig. S4B and S4C).
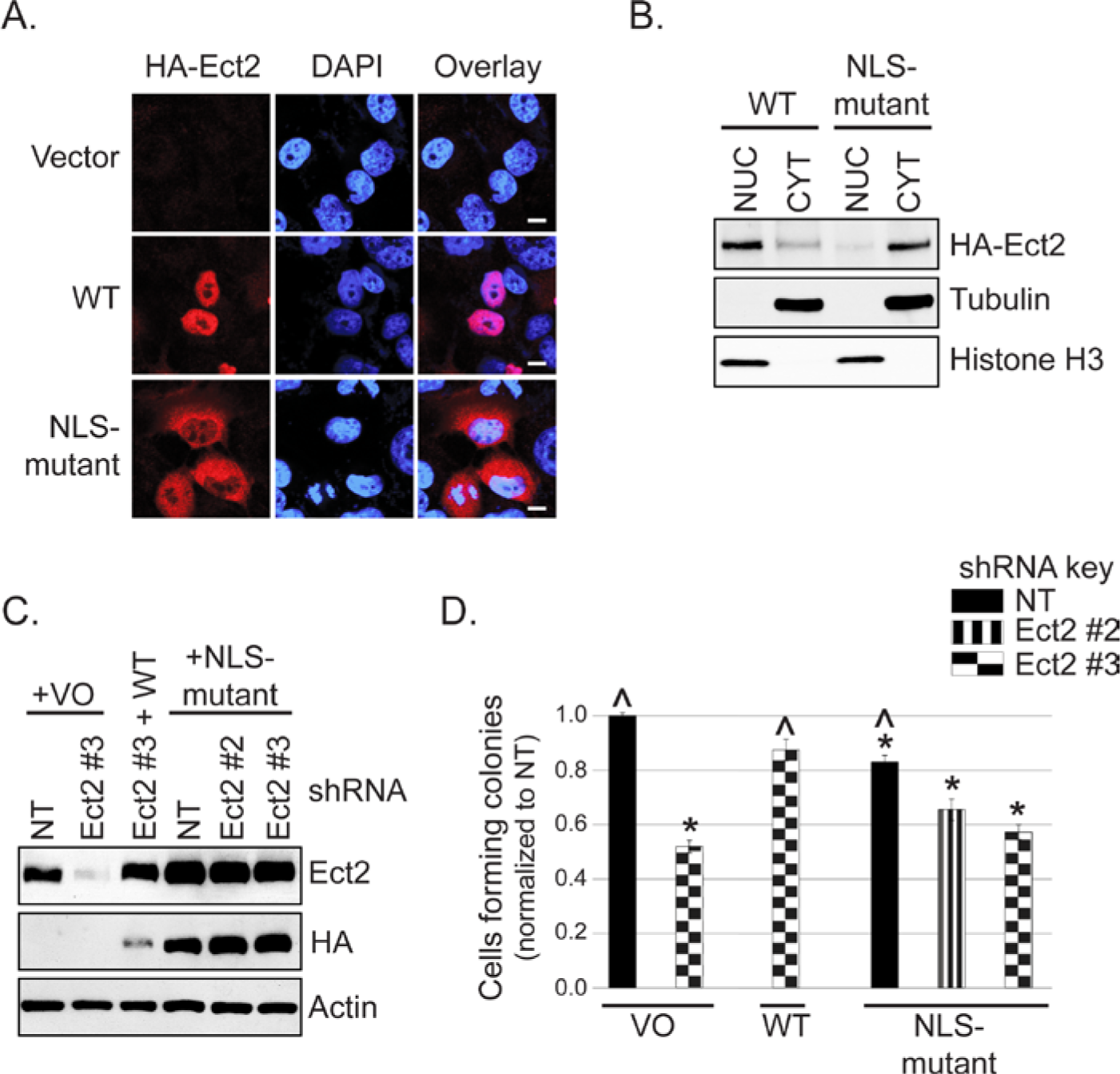
Predominantly nuclear localization is required for Ect2-mediated transformed growth. (
Despite higher levels of expression than WT Ect2 (Fig. 5C), the NLS-mutant was not capable of rescuing the anchorage-independent growth defect of Ect2 knockdown cells (Fig. 5D). Interestingly, its overexpression alone in cells retaining endogenous Ect2 was sufficient to lead to decreased anchorage-independent growth. This suggests that simply localizing Ect2 to the cytoplasm in the presence of nuclear Ect2 is not sufficient to drive transformation in these ovarian cancer cells, but rather may have the opposite effect, consistent with our TMA findings in which higher levels of cytoplasmic Ect2 were found preferentially in the less-advanced lesions.
Some nuclear Ect2 is present in an active conformation that has enhanced specificity for Rac1
It has been proposed that nuclear Ect2 is auto-inhibited,8,37 whereas our results suggest at least a portion is active. To test this directly, we fractionated OVCAR8 cells and performed a pulldown assay38,39 for active Ect2 using a constitutively nucleotide-free form of RhoA(17A), which binds with high affinity to the active form of RhoGEFs. 39 Using tubulin as a cytoplasmic marker and PARP as a nuclear marker, we confirmed effective fractionation (Fig. 6A, left). Immunoblotting of endogenous Ect2 demonstrated that, while the vast majority was observably nuclear (NUC), some Ect2 was detectable in the non-nuclear fraction (CYT). This is unlikely to be due to nuclear contamination of the CYT fraction because PARP was not detectable there. In both fractions, Ect2 bound with notably higher affinity to GST-RhoA(17A) compared to the GST-only control (Fig. 6A, right), revealing that some active Ect2 is present in both compartments.
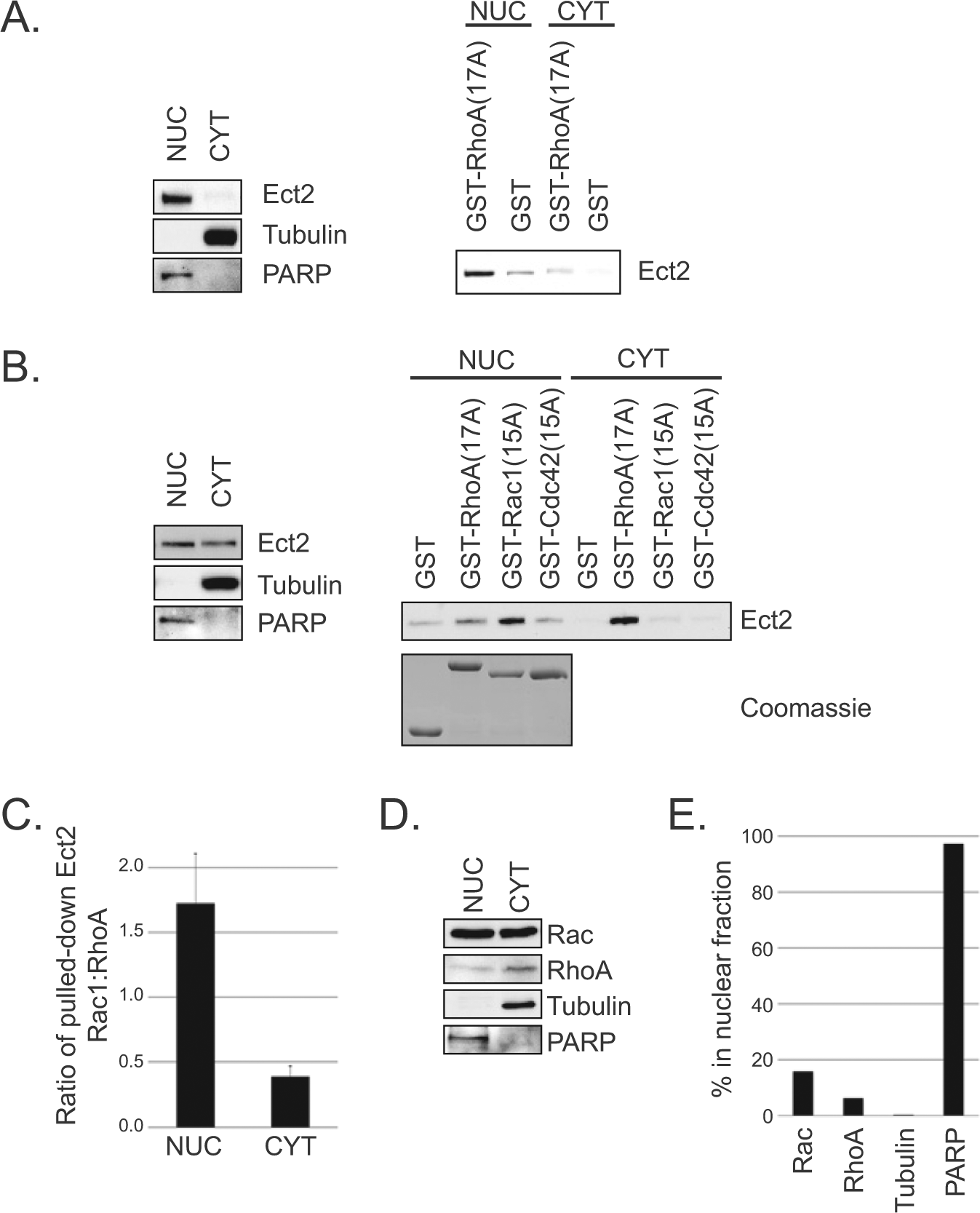
Some nuclear Ect2 is present in an active conformation that has enhanced specificity for Rac. (
The ability of a GEF to interact with nucleotide-free mutant GTPases is dependent on both the concentration of active GEF 38 and the specificity of the GEF for the GTPase.39-41 To determine the ability of Ect2 to interact with other Rho GTPases within each subcellular compartment, we extended these results with GST-RhoA(17A), GST-Rac1(15A), and GST-Cdc42(15A). Equal amounts of GST-Rho proteins were added in excess to each Ect2-containing lysate (Fig. 6B, Coomassie), so the relative amounts of endogenous RhoA and Rac in each cellular compartment were irrelevant and could not contribute to any observed differences in Ect2 interactions. Additionally, to avoid complications from the vastly different amounts of Ect2 present in the nuclear versus non-nuclear compartments, here we pulled down Ect2 from lysates containing equal amounts of total Ect2 rather than equal amounts of total protein.
Unexpectedly, the Rho GTPase interactions of Ect2 in the nucleus were different from those in the cytoplasm (Fig. 6B). In the nucleus, Ect2 interacted with both Rac1(15A) and RhoA(17A), with a distinct preference for Rac1 despite equal exposure to both. In the cytoplasm, Ect2 interacted almost exclusively with RhoA. The minimal levels of Ect2 pulled down with Cdc42(15A) from either compartment are consistent with our proposal that activation of Cdc42 via Ect2 is indirect in these cells. Using densitometry to quantify the amount of Ect2 from each compartment that was pulled down by Rac versus Rho, we calculated that the ratio of Ect2 pulled down by Rac to that of Ect2 pulled down by Rho was 1.7 in the nucleus but only 0.4 in the non-nuclear compartment (Fig. 6C). Because equal amounts of ectopic Rac and Rho were available for Ect2 binding in each case, these different ratios suggest the possibility of an intrinsic conformational difference between nuclear and cytoplasmic Ect2.
We detected both endogenous Rac and endogenous Rho in the nucleus (Fig. 6D), but with unequal distribution: a greater proportion of Rac was distributed to the nucleus (16%) than was Rho (6%) (Fig. 6E). Thus, intrinsic differences detected by the pulldowns using ectopic nucleotide-free GTPases are likely to be amplified by the relative availability of endogenous Rac and Rho in each compartment during cellular signaling.
Ect2 activates endogenous Rac in the nucleus and endogenous RhoA in the cytoplasm
To determine if Ect2 is capable of activating endogenous Rac and/or Rho in the nucleus, we first performed standard Rho binding domain (RBD) recruitment assays.42-46 We used POSH, a downstream effector specific to Rac, 47 to detect active Rac-GTP. Under basal conditions, GFP-POSH-RBD was expressed diffusely throughout the cytoplasm and nucleus, but was recruited to the nucleus in the presence of exogenous WT Ect2 (Fig. 7A; quantified in Fig. 7C). Nucleolar exclusion of GFP-POSH-RBD was evident by confocal microscopy (Fig. 7A); thus, GFP-POSH-RBD was recruited into the nucleus and not onto the nuclear envelope. Recruitment was dependent on both GEF activity and nuclear localization of Ect2, because the GEF-deficient and the NLS-mutant each failed to recapitulate the effects of WT Ect2. Consistent with these results, GFP-PAK1-RBD 48 was also recruited to the nucleus by Ect2, in a GEF- and NLS-dependent manner (Suppl. Fig. S5A and S5B). These results indicate that WT Ect2 activates endogenous Rac1, and the site of activation is primarily in the nucleus.
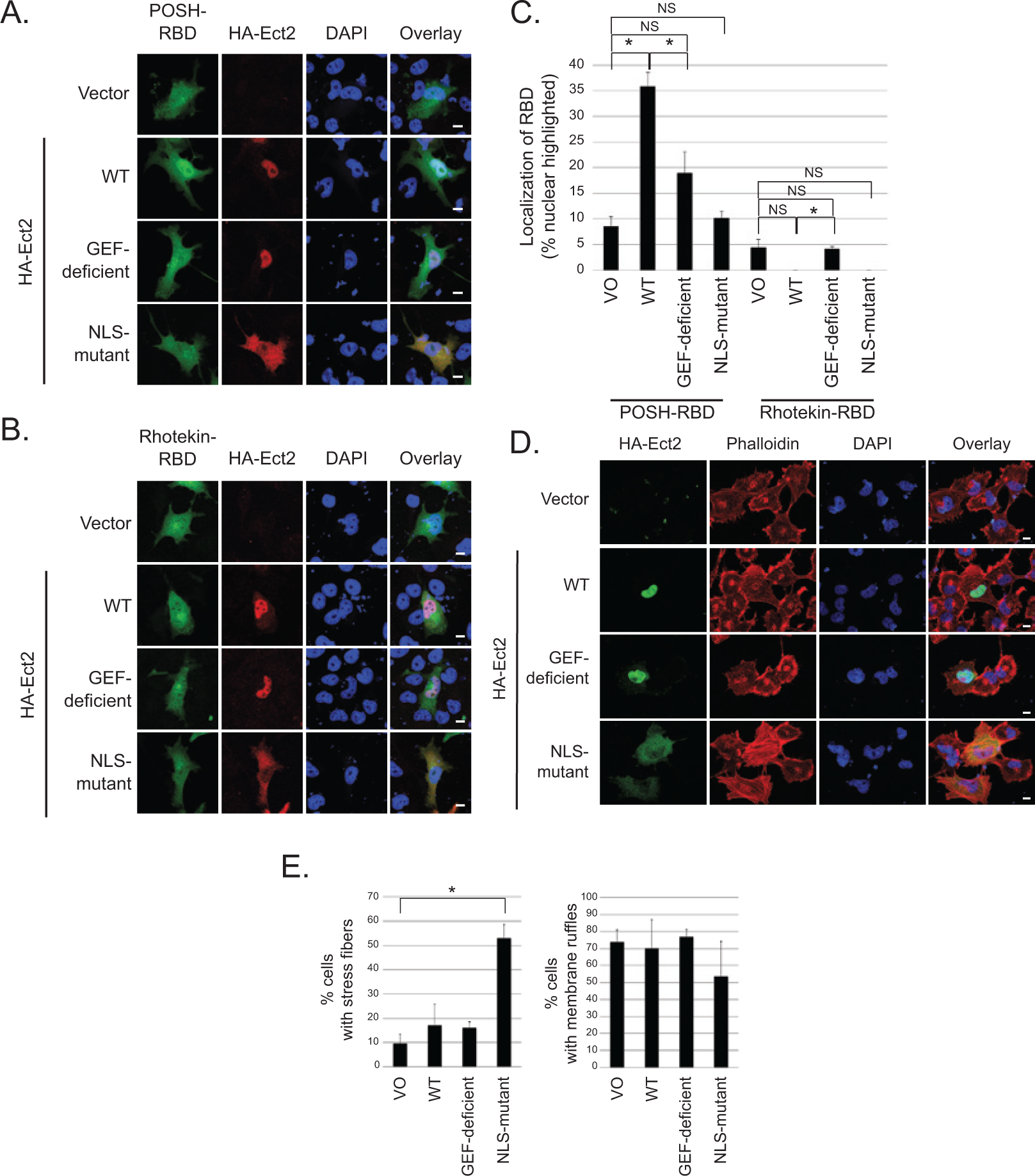
Ect2 recruits downstream effectors of Rac to the nucleus and initiates canonical RhoA signaling in the cytoplasm. To determine if Ect2 was capable of activating endogenous Rho GTPases in the nucleus, we examined GFP-RBD recruitment in OVCAR8 cells expressing empty vector versus exogenous HA-tagged WT, GEF-deficient, or NLS-mutant Ect2. Confocal immunofluorescence microscopy was used to image cells expressing both HA-Ect2 (red) and each GFP-RBD (green); nuclei were stained using DAPI (blue). (
Notably, the cytoplasmically localized NLS-mutant did not activate Rac as measured either by recruitment assay (Fig. 7A, Suppl. Fig. S5A) or by pulldown assay (Fig. 4A). In contrast, we observed that the cytoplasmically localized NLS-mutant Ect2 strongly increased RhoA-GTP levels (50.5-fold, Fig. 4A). These results indicate that Ect2 activity in the cytoplasm preferentially results in activation of Rho. However, GFP-Rhotekin-RBD, a surrogate for RhoA activity, 49 was not recruited either to the cytoplasm or to the nucleus (Fig. 7B, quantified in Fig. 7C). One reason for this may be that any shift of the smaller pool of nuclear GFP-Rhotekin-RBD into the cytoplasm may not be detectable in the larger pool of cytoplasmic Rhotekin-RBD. Therefore, to determine if the elevated RhoA-GTP that we detected by pulldown led to a biological consequence in the cytoplasm, we performed phalloidin staining to examine the actin cytoskeleton of OVCAR8 cells exogenously expressing WT or mutant Ect2. The canonical cytoskeletal consequence of RhoA activity is the formation of stress fibers, 50 and indeed we found that only the cytoplasmically localized NLS-mutant Ect2 significantly enhanced stress fiber formation, whereas the nuclearly localized WT and GEF-deficient Ect2 did not (Fig. 7D and E). These results indicate that cytoplasmically localized Ect2 can functionally activate RhoA. In contrast, and consistent with its inability to activate Rac (Fig. 4A), NLS-mutant Ect2 did not increase membrane ruffling, a canonical consequence of Rac activity 51 (Fig. 7D and E). WT Ect2 also did not enhance membrane ruffling, consistent with a previous report that overexpression of a nuclearly localized, constitutively active Rac1 mutant enhanced cell proliferation without inducing membrane ruffles. 34 Collectively, our results indicate that Ect2 preferentially activates Rac1 in the nucleus and RhoA in the cytoplasm. The larger pool of cytoplasmic Rho family GTPases (Fig. 6E) may explain why we observed more striking changes in RhoA activation compared to Rac activation upon Ect2 overexpression (Fig. 4A) and knockdown (Fig. 4D) in whole cell lysate.
Nuclear Rac1 activity is sufficient to rescue defects in Ect2-mediated transformed growth
Since we have demonstrated that both nuclear localization and GEF activity are required for Ect2-mediated transformed growth in ovarian cancer cell lines and that Ect2 preferentially activates Rac1 in the nucleus, our results suggest that Ect2 activation of nuclear Rac may be an important contributor to its transforming ability.
We first attempted to determine directly whether Rac1 is necessary for Ect2-driven anchorage-independent growth by asking whether Rac1 knockdown impaired colony formation in soft agar. However, it was impossible to generate cells lacking Rac1, as transfection with any of 4 independent Rac1-targeted siRNAs (Dharmacon/Thermo Scientific, Pittsburgh, PA) induced widespread cell death (data not shown), leaving us with insufficient Rac1-knockdown cells for analysis and indicating that Rac1 is an essential protein for viability of these cells. Although it is likely that the non-nuclear functions of Rac1 were critical, we were unable to selectively knock down only nuclear Rac1.
Another approach to testing our model is to determine whether nuclear Rac1 can overcome the defect in transformed growth conferred by loss of Ect2 expression. To test this, we generated HA-tagged activated Rac1 mutants that were preferentially localized in the nucleus (Fig. 8A). We used mutants of Rac1 that were activated independently of Ect2 by either the traditional GTP-locked G12V mutation 51 or by the fast-cycling F28L mutation. 52 Prenylation of Rac1 is required for its plasma membrane association 5 and impairs its nuclear import. Non-prenylated Rac1 localizes strongly to the nucleus when GFP-tagged.34,53 However, we determined that an additional N-terminal NLS was required to concentrate unprenylated HA-tagged Rac1 in the nucleus. Therefore, to preferentially localize the activated Rac1 mutants to the nucleus, we both mutated the Rac1 prenylated cysteine to serine (C189S, “SAAX” mutation) and added the classical NLS from SV40T antigen 54 to the N-terminus to generate “NLS-Rac1-SAAX” mutants. When expressed stably in cells with Ect2 knocked down (Fig. 8B), each of these nuclearly concentrated, active mutants of Rac1 was able to rescue anchorage-independent growth to the same extent as re-expression of WT Ect2 (Fig. 8C). These results support our model that Ect2 activation of Rac1 in the nucleus is a key component of its transforming function.
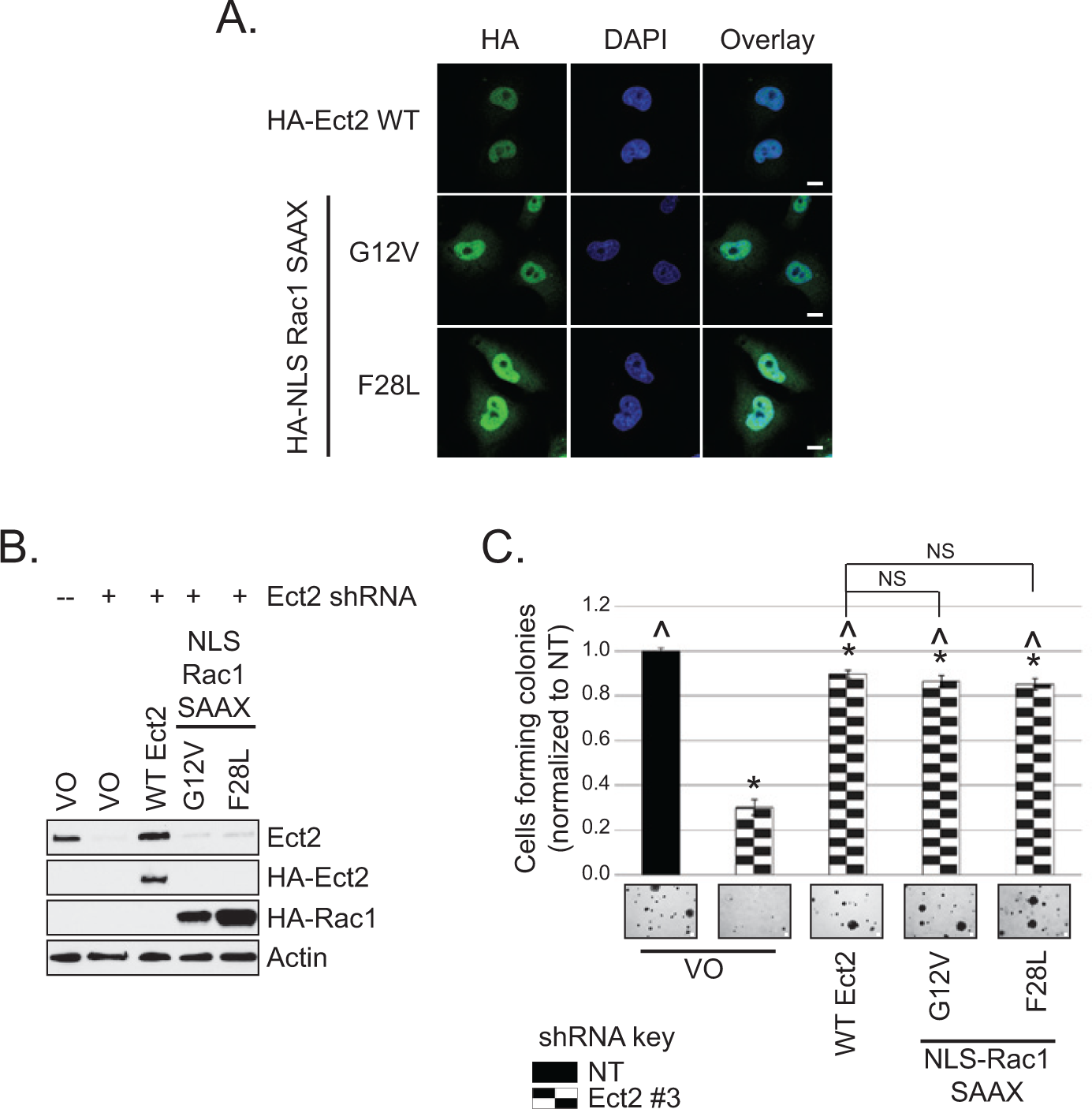
Nuclear Rac1 activity is sufficient to rescue defects in Ect2-mediated transformed growth. (
Discussion
Despite the largely nuclear localization of the RhoGEF Ect2 in interphase cells,7,13,15,55,56 previous studies have focused on its actions at non-nuclear locations. For example, the most prominent role for Ect2 in normal cells is its requirement during cytokinesis, when the nucleus is not intact and Ect2 activates RhoA at the cleavage furrow and midbody.7,10,11,25,57 But the function of Ect2 in cytokinesis of normal cells can be uncoupled from its role in promoting transformed growth of tumor cells,11,15,58 leaving the mechanisms whereby it promotes transformation less well understood. One model suggests that Ect2 must be cytoplasmically mislocalized to drive transformation.8,15 In contrast, our study indicates a prominent role for a nuclear pool of Ect2 in the transformed phenotype of ovarian cancer cells. Furthermore, we have identified a requirement for Ect2 to act as a RhoGEF within the nucleus to promote growth transformation in ovarian cancer cells.
In both ovarian cancer patient tumor tissues and established cell lines, we found that Ect2 protein was expressed robustly, consistent with its published genomic amplification and mRNA overexpression in ovarian tumors.22,24,25 Ect2 was predominantly localized to the nucleus, particularly in advanced disease. Interestingly, we did not observe significant Ect2 localization to the cytoplasm in advanced tumors; instead, benign ovarian cysts had the most prominent cytoplasmic staining. Although Ect2 was reported to be strongly mislocalized from the nucleus to the cytoplasm in non–small cell lung cancers (NSCLC) compared to normal lung, 15 our observations of increased nuclear expression in ovarian cancers are more consistent with other reports where inspection of the data shows that Ect2 increases globally on tumor progression, with extensive increases in nuclear expression and less dramatic increases in cytoplasmic Ect2.13,56,59,60 We show here that both Ect2 nuclear localization and its RhoGEF activity are required for Ect2-driven anchorage-independent growth of ovarian cancer cells, suggesting that Ect2 acts as a GEF from within the nucleus to drive transformation. Indeed, our observations that cytoplasmic Ect2 localization was inversely correlated with advanced disease, and that a cytoplasmically localized Ect2 NLS-mutant reduced anchorage-independent growth, indicate that mislocalization of Ect2 to the cytoplasm does not drive ovarian transformation but rather might oppose the effect of nuclear Ect2. That this NLS-mutant Ect2 was able to act as a GEF to increase RhoA-GTP and promote stress fiber formation suggest the intriguing possibility that Ect2 activation of RhoA in the cytoplasm might even counteract its actions in the nucleus.
In NSCLC cell lines, transformed growth can be driven by Ect2-induced activation of Rac1. 15 Rac1 activation was proposed to occur in the cytoplasm, although this was not tested directly.15,58 Since the BRCT domains of Ect2 can auto-inhibit its activity,8,37,61 nuclear Ect2 was proposed to be auto-inhibited and sequestered from Rho GTPases. 8 Here, we found that active Ect2 was present in the nucleus. There has been almost no prior investigation of the subcellular distribution of active Ect2, but one study did identify active Ect2 in the nucleus of HEK293 cells. 36
While Rho GTPases are described as cytoplasmic proteins, 5 this is not the entire story. In particular, Rac132-34 and, to a lesser extent, RhoA35,36 and Cdc42 62 have all been detected within the nucleus. We were able to detect both endogenous Rac and some endogenous RhoA in the nucleus of OVCAR8 cells; therefore, both are available in the nucleus as substrates for Ect2 RhoGEF activity. Our work also provides further evidence for nuclear activation of Rho GTPases. Although early reports simply described nucleocytoplasmic shuttling,32,33 there are recent reports that RhoA can be activated from within the nucleus in some contexts.35,36
We observed that Ect2 preferentially activates Rac in the nucleus. We also identified at least 2 likely contributors to this finding: (a) the presence of more Rac than RhoA in the nuclear compartment and (b) an intrinsic substrate specificity, such that nuclear Ect2 preferentially binds to Rac1 whereas cytoplasmic Ect2 preferentially binds to RhoA. Consistent with the latter, the NLS-mutant, which is defective for nuclear localization and is distributed well into the cytoplasm, robustly activated RhoA but not Rac.
Modulation of intrinsic substrate specificity may be complex. Phosphorylation is required to optimally activate Rac both in vitro 7 and in cells. 63 We have observed that bacterially expressed, that is, unphosphorylated Ect2 can activate RhoA but not Rac1 in vitro. 57 It is tempting to speculate that nuclear Ect2 is appropriately phosphorylated; however, despite repeated attempts we have not observed mobility differences between nuclear and cytoplasmic Ect2 (as observed previously for phosphorylated versus unphosphorylated Ect27,10). Alternatively, as-yet-undefined protein-protein interactions that occur in the nucleus may promote interaction with Rac, perhaps involving the Ect2 C-terminus, which is required to activate Rac in cells. 64 Similarly, cytoplasmic scaffolding may promote RhoA but not Rac activation. Protein-protein interactions have been speculated to explain reports of varying Ect2 specificity throughout cytokinesis/mitosis. 65 Regardless of the mechanism, to our knowledge, this study is the first to demonstrate altered specificity of a RhoGEF based on its subcellular localization.
Our work is also the first to show that active Rac1 is capable of driving transformation when concentrated in the nucleus (and excluded from the plasma membrane). The precise function of nuclear Rac in these cells is unknown. Forced expression of a constitutively active but unprenylated Rac1 mutant in the nucleus of NIH3T3 cells accelerated cell cycle progression. 34 However, this mutant did not induce lamellipodia formation, a primary function of cytoplasmic Rac1, 34 suggesting that nuclear and cytoplasmic functions of active Rac may be distinct. Similarly, although overexpression of Ect2 was capable of recruiting the RBD of Rac effectors to the nucleus, we did not observe an increase in lamellipodia or membrane ruffling on expression of Ect2. Still, negative regulators66,67 and downstream signaling molecules68-72 of Rac can be detected in the nucleus, suggesting that some cytoplasmic signaling pathways could remain intact there, albeit with distinct stoichiometry. Some Rac effectors directly regulate transcription from the nucleus,68-70 but we did not observe Ect2-mediated alterations in transcriptional activation (data not shown). The speckled pattern of Ect2 localization in the nucleus suggests that Ect2 localizes to distinct subnuclear structures. Nuclear speckles regulate mRNA splicing, 73 and Ect2 interacts with the SNRNP200 gene product, a component of the spliceosome, 74 so it is possible that Ect2 signaling to Rac plays a role in this process.
In conclusion, here we identified Ect2 as necessary for cellular transformation in ovarian tumor cells. We also identified a novel mechanism through which Ect2 can drive cellular transformation by acting as a GEF from within the nucleus, in a manner distinct from its actions in the cytoplasm. We showed that Ect2 preferentially activates Rac in the nucleus of ovarian cancer cells and RhoA in the cytoplasm. In addition to Ect2, the RhoGEFs Net1, LARG, and Vav3 have also been identified in the nucleus.75-77 It would be interesting to determine if subcellular localization also alters the specificity of these and other RhoGEFs. Our results suggest that nuclear Rac1 activity is biologically relevant for transformed growth and implicate activation of nuclear Rac as a mediator of Ect2-driven transformation. In the future it may become possible to selectively ablate Ect2-activated nuclear Rac. If so, this would allow more rigorous testing of a requirement for nuclear Rac activity in Ect2-driven ovarian cancer cell transformation. As physiologically relevant substrates of nuclear Ect2 and Rac1 are identified, it will also be interesting to uncover their roles in transformation.
Materials and Methods
Molecular constructs and transfections
Short hairpin RNA (shRNA) against human Ect2, non-targeting (NT) shRNA, and shRNA-insensitive full length Ect2 were generously provided by Alan Fields (Mayo Clinic, FL). 15 Quikchange II site-directed mutagenesis kit (Stratagene, Santa Clara, CA) was used to create NLS- and DH-mutant Ect2 from shRNA-insensitive full length Ect2 (primers, Suppl. Fig. S6). NLS-Rac1-SAAX mutants were generated using PCR (primers, Suppl. Fig. S6) to amplify Rac1 F28L 57 and Rac1 G12V (gift of Peter Hordijk, Sanquin). All Ect2 and Rac1 expression constructs were cloned into pFugW-HA-blasticidin (gift of Jeran Stratford, UNC-CH). Viral psPAX2 (packaging) and pMDG.2 (envelope) vectors were from Addgene. pGEX-2T and RBD constructs45,78,79 were gifts from Keith Burridge (UNC-CH). Transfections were performed using TransIT-LTI (Mirus, Madison, WI) according to the manufacturer’s instructions.
Cell culture, lentiviral infection, and generation of stable transfectants
Human ovarian epithelial cancer cell lines were grown in RPMI-1640 with 10% fetal bovine serum (FBS; Sigma, St. Louis, MO). 293T cells were grown in DMEM-H + 10% FBS. Lentiviruses were generated as described, 80 except virus was collected 24 and 48 hours posttransfection. Lentivirus-infected cells 80 were selected in puromycin (1 µg/mL, Cellgro, Manassas, VA) or blasticidin (10 µg/mL, Invitrogen, Grand Island, NY).
Flow cytometry
Flow cytometry was performed exactly as we have described previously. 57
Immunoblotting, immunofluorescence, phalloidin staining, and microscopy
Immunoblotting was performed from RIPA cell lysates by our standard methods. 45 For immunofluorescence and phalloidin staining, cells were plated on fibronectin, fixed in paraformaldehyde, exposed to antibodies or to Alexa Fluor568-conjugated phalloidin (Invitrogen), and confocal microscopy imaging performed as we have done previously.45,81 A Nikon (Melville, NY) Eclipse TS100 microscope was used to image cells and colonies in bright field.
Ovarian tumor tissue microarray (TMA) immunohistochemistry
A previously validated TMA,26-29 containing formalin-fixed, paraffin-embedded primary epithelial ovarian carcinomas or benign cysts, was stained for Ect2 by the UNC-Translational Pathology Laboratory Core. We used a standard method of scoring the TMA, 29 except each core was scored separately for nuclear and cytoplasmic staining. Each compartment was scored separately on a scale of 0 to 100 for percent cyst or tumor cells stained, and on a scale of 0 to 3 for average intensity of staining, where 0 was no staining and 3 was maximal staining. The score for each compartment was the product of percent cells staining in the nucleus/cytoplasm multiplied by the average intensity of nuclear/cytoplasmic staining for each core.
Previously collected patient data26-29 allowed us to classify the serous tumors by degree of malignancy (cysts, serous borderline tumors, serous low-grade tumors, and serous high-grade tumors). A Kruskal-Wallis test was performed to compare both nuclear and cytoplasmic scores among these groups; P < 0.05 was considered significant. Institutional review board approval was through UNC-CH IRB 08-0242.
Anchorage-dependent and -independent growth assays
Soft agar assays to assess anchorage-independent growth were performed and counted as we have described. 45 A 2-tailed Student’s t-test assuming unequal variance was performed on colony counts. The P-values were adjusted for multiple tests using the Bonferroni correction, and P < 0.05 was considered significant. Standard MTT proliferation assays were performed. 82 Cells were normalized to the average value for day 0 and growth curves were generated from the average of all experiments. Slopes of the log-transformed growth curves were compared using likelihood ratio tests to determine statistical significance, and a Bonferroni corrected P-value <0.05 was considered significant.
Cell fractionation
Cell fractionation into nuclear and postnuclear compartments was performed by hypotonic swelling, Dounce homogenization, centrifugation through 28.5% iodixanol (Sigma), and sonication as described in detail previously. 38
Pulldown and recruitment assays for active Ect2 and Rho GTPases
Active Ect2 was assessed by incubating fractionated cell lysates with nucleotide-free Rho GTPases isolated from bacteria and bound to agarose beads, resolved on SDS-PAGE, and immunoblotted for Ect2.38,39 Prior to performing pulldowns, fractions were subjected to Bradford analysis for total protein content. For some experiments (Fig. 6A), equal amounts of total protein were added to the beads. Alternatively (Fig. 6B), loading of fractionated cell lysates was normalized for equal expression of Ect2. This was done by comparing the distribution of Ect2 in nuclear versus cytosolic fractions using densitometry of western blots of the fractionated lysates, which showed that loading 2.6× more of the cytosolic fraction resulted in approximately equal loading of Ect2 from each fraction. Loading of Ect2 was examined by western blot following each of these assays. Beads were blocked in 5% BSA for 30 minutes to minimize nonspecific binding of Ect2 to GST. Equal loading of the beads was confirmed by SDS-PAGE and Coomassie blue staining.
To determine levels of active, GTP-bound RhoA, Rac, and Cdc42, pulldowns using GST-Rhotekin-RBD or -PAK-PBD, respectively, 78 were performed from whole cell lysates. Densitometry on immunoblots was performed using ImageJ and normalized as described in the Results section.
Recruitment assays42-46 using GFP-tagged RBDs for Rhotekin, POSH, and PAK were used to monitor changes in the subcellular distribution of active endogenous Rho GTPases. Fixed cells expressing HA-Ect2 in each specific location (vector: diffuse; WT and DH-mutant: nucleus; NLS-mutant: cytoplasm) that co-expressed GFP-RBD were scored for GFP-RBD localization based on the following categories: nuclear-excluded, plasma membrane-highlighted, diffuse nuclear and cytoplasmic localization, nuclear-highlighted. The average percentage of cells nuclear-highlighted out of total cells counted is shown. Statistical significance was determined as described for the soft agar assays.
Footnotes
Acknowledgements
We thank Kent Rossman for advice on designing the GEF-deficient Ect2 mutant; Alan Fields for the gift of Ect2 plasmids; Keith Burridge for the gift of RBD plasmids and helpful advice; Marc Olorvida of the UNC-Translational Pathology Core Laboratory for assistance with immunohistochemistry; Robert Bagnell of the UNC-Microscopy Services Laboratory for assistance with microscopy; Rafael Garcia-Mata for advice on fractionation-pulldown assays; and Jim Fiordalisi for assistance with plasmid constructs.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (Grant Numbers CA109550, CA042978, F31-CA165844, T32-GM00 7040, F31-CA159821, T32-CA71341), the Howard Hughes Medical Institute (Med-Into-Grad Program), and the Office of Undergraduate Research at the University of North Carolina at Chapel Hill (Summer Undergraduate Research Fellowship).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.