
Abstract
The activity of c-Jun N-terminal kinase (JNK) was initially described as ultraviolet- and oncogene-induced kinase activity on c-Jun. Shortly after this initial discovery, JNK activation was reported for a wider variety of DNA-damaging agents, including γ-irradiation and chemotherapeutic compounds. As the DNA damage response mechanisms were progressively uncovered, the mechanisms governing the activation of JNK upon genotoxic stresses became better understood. In particular, a recent set of papers links the physical breakage in DNA, the activation of the transcription factor NF-κB, the secretion of TNF-α, and an autocrine activation of the JNK pathway. In this review, we will focus on the pathway that is initiated by a physical break in the DNA helix, leading to JNK activation and the resultant cellular consequences. The implications of these findings will be discussed in the context of cancer therapy with DNA-damaging agents.
Keywords
Introduction
In physiological conditions, eukaryote genomes are subjected to a variety of DNA-damaging events that may alter their coding information. Double-strand breaks (DSBs) of the DNA double helix are the most deleterious events that occur in the genome and can either be due to endogenous causes such as the metabolic production of reactive oxygen species (ROS) and replication fork collapse or exogenous causes including exposure to chemical genotoxic agents or ultraviolet (UV) irradiation. In order to maintain genome homeostasis, eukaryotes have evolved protective mechanisms that can detect damaged DNA, repair it and induce an appropriate cellular response including JNK signaling activation.1-7 of DSBs seems to rely on the initial detection of chromatin remodeling induced by the breakage that initiates a signaling cascade to activate repair systems. The 2 major mechanisms responsible for eliminating DSBs are the well-described nonhomologous end joining and homologous recombination repair systems, considered, respectively, as a fast and a slow repair system. The means of activation of these repair pathways are well known, and the first event described is the formation of nuclear foci, called irradiation-induced foci (IRIF), which contain a variety of proteins including the phosphorylated histone H2AX (γH2AX) that is common to every DNA damage response (DDR) described so far.8,9 When these mechanisms fail to repair damaged DNA, the persistence of the signal emerging from broken DNA leads to cell cycle arrest, apoptosis, or cell senescence. In cells compromised for cell cycle checkpoints, the persistence of damage in the genome during mitosis can lead to mutations or the missense recombination of chromatids and cell transformation. An initial observation has linked DNA damage to the activation of NF-κB, but the mechanism responsible for driving the nuclear DNA damage signal to the NF-κB cytoplasmic target has only recently been extensively described. The NF-κB family is a group of 5 transcription factors that bind DNA as dimers (namely RelA, RelB, c-Rel, p50, and p52). These transcription factors were initially discovered as inflammation-activated factors, but they can also be activated by a wide variety of signals including bacterial infection, presence of inflammatory cytokines, or DNA-damaging agents. The inactive form of NF-κB is sequestered in the cytoplasm through binding to its IκB inhibitory partner. NF-κB–activating signals induce the phosphorylation of IκB by a 3-subunit complex called IκB kinase (IKK) that is composed of at least 2 catalytically active kinases (IKKα and IKKβ) and a regulatory subunit, the NF-κB essential modifier (NEMO). 10 In response to its phosphorylation, IκB undergoes ubiquitin-mediated proteasomal degradation, and the released NF-κB dimers can translocate to the nucleus and interfere with their target sequences. Although the observation that UV-induced DNA damage can activate NF-κB signaling was made a long time ago, the mechanisms responsible for driving the nuclear DNA damage signal to the NF-κB cytoplasmic target has only recently been extensively described.11,12 Mechanistic insight of JNK activation induced by DSB was shown by the discovery of DDR-driven activation of NF-κB and the consecutive production of tumor necrosis factor α (TNF-α) responsible for an autocrine feed-forward signaling loop through its type 1 receptor (TNFR-1) and the consequent activation of the JNK signaling pathway. 13
DNA Damage Forward Signaling
Initiation of the cellular response to genotoxic stress relies on the efficient detection of any DNA lesion in the whole genome. While the events occurring shortly after DNA breakage are well described, the initial signal responsible for triggering these events has not yet been clearly identified. The proposed model for explaining the detection of DSBs stipulates that when a DSB occurs, the topological constraints due to DNA supercoiling are released, which induces the relaxation of the chromatin fiber.14-16 The associated conformational changes are proposed to recruit the wide variety of factors responsible for posttranslational modifications of the DNA- associated proteins. The earliest event in this process is histone poly (ADP- ribosylation), during which time enzymes called the poly (ADP-ribose) polymerases (PARP1, PARP2, and PARP3) catalyze the formation of poly (ADP-ribose) (PAR) and their covalent linkage to lysines in core histone proteins.9,17,18 Notably, the PAR chains formed by PARP1 serve as a binding site for chromatin and histone modifiers that inhibit replication and transcription around the site of the lesions. 19 Very early after formation of the DNA damage, γH2AX is also detectable in close vicinity to the DSBs as well as other components of the DNA repair pathways that accumulate in so-called IRIF. 20 Phosphorylation of histone H2AX is performed by kinases of the phosphoinositide-3-kinase–related protein (PIKK) family called DNA-dependent protein kinase (DNA-PK) and ataxia telangiectasia mutated (ATM). 21 DNA-PK constitutes a DNA-binding subunit (Ku70 and Ku80) responsible for localization of the kinase subunit (DNA-PKcs) at DNA free ends. 22 Although this composition would be a good candidate for signaling downstream events from DSBs, the current general consensus is that it does not play a central role in this phenomenon. Instead, while the intermediates responsible for ATM localization at DSBs are not yet well described, ATM has been shown to be activated through transautophosphorylation-induced dissociation of ATM-inactive dimers.17,23 Active ATM is shown to be prominently involved in the phosphorylation of H2AX and spreading of DSB signaling at the distance of IRIF.9,24,25 In turn, γH2AX, together with a factor called mediator of DNA checkpoint 1 (MDC1), was shown to be necessary for the recruitment of key elements involved in DSB signaling such as 53BP1, BRCA1, and p53 tumor suppressor protein.26,27 In consequence of these initial nuclear events, DDR signaling is transduced to cytoplasmic targets, among which NF-κB and the JNK pathway are prominently involved in the proliferation and survival of the cells.
From DNA Damage to NF-κB and JNK Activation
The demonstration that NF-κB activation can be induced by UV exposure suggested that it could be the mechanism responsible for triggering the DDR. Apparent contradictory results initiated a debate over whether UV-induced NF-κB activation is directly dependent on DNA damage signaling or not.28-30 The explanation seems to lie in the middle, with signals directly emanating from the nucleus due to the activation of DNA damage pathways combined with other events such as UV-induced formation of reactive species converging to activate NF-κB.31,32 Major breakthroughs confirming the existence of a nuclear-to-cytoplasmic signal transduction came from Miyamoto’s team, who broadly deciphered the mechanisms governing ATM- and NEMO-dependent nucleus-to-cytoplasm signaling responsible for NF-κB activation.33-36 A fundamental development confirming the existence of a nucleus-to-cytoplasm signaling pathway came from extending the results obtained with UV radiation activation of NF-κB to a variety of other DNA-damaging agents such as etoposide or camptothecin.36,37 Another convincing line of evidence comes from an experiment using Atm–/– mice: while control mice show the activation of IKK in response to whole body irradiation, Atm–/– mice fail to activate NF-κB signaling, demonstrating the requirement of ATM for transduction of the nuclear DNA damage–driven activation of NF-κB. 38 Initial studies described the sumoylation of NEMO by the small ubiquitin-related modulator 1 (SUMO-1) as the driver for nuclear relocalization of IKK-unbound cytoplasmic NEMO proteins.33-39 While this sumoylation event was described to be ATM independent, the complete activation of NF-κB upon genotoxic damage also relies on nuclear NEMO posttranslational modifications, resulting in the export of an active form of NEMO. One of the earliest and critical events in DNA damage recognition and signaling is PAR formation by PARP-1, which has been demonstrated to play a fundamental role in the mechanism leading to NEMO nuclear posttranslational modifications. 40 The automodification of PARP-1 was shown to drive the assembly of a complex consisting of ATM, NEMO, and the sumoylation factor inhibitor of activated STAT γ (PIASγ). Formation of this complex is concomitant with posttranslational modifications of NEMO that appear to be necessary for DNA damage–induced NF-κB activation: PIASγ transiently sumoylates NEMO on lysines 277 and 309, and ATM phosphorylates NEMO on serine S85.34,35 These initial modifications are required for the subsequent ubiquitination of NEMO by the protein cIAP1 (an inhibitor of apoptosis), and this modification is proposed to drive the nuclear export of a complex comprising at least NEMO and ATM. 33 The multimodal interactions occurring in the nucleus and the cytoplasm between NEMO and its posttranslational modifiers appear to be partly dependent on the cell type and extent of the DNA damage. However, a common cytoplasmic effector of IKK activation by DNA damage is transforming growth factor β–activated kinase 1 (TAK1), a kinase that has already been shown to be an activator of NF-κB in other cellular contexts. 41 Although the proposed models tend to differ depending on the cell type used, TAK1 was shown to be required for genotoxic-induced activation of NF-κB.35,42 Upon DNA damage– mediated activation of ATM and NEMO, TAK1 is activated through the ubiquitination of its regulatory partners and in turn phosphorylates IKKβ, which leads to the activation of NF-κB.13-35 The demonstration that UV-challenged IKKβ knockout cells lack the degradation of IκB and activation of NF-κB strongly implicates IKK involvement in DNA damage–induced activation of NF-κB.35,42,43 Recent studies also suggest that NF-κB activation can occur independently of IKK catalytic activity upon DNA damage. Using nonphosphorylatable forms of IκB, Tsuchiya and colleagues 44 demonstrated that the IKK complex can play the role of an adaptor protein responsible for targeting IκB to ubiquitination independently of phosphorylation. Another mode of NF-κB activation by UV radiation was also suggested to occur via the DNA damage–induced phosphorylation of eukaryotic initiation factor 2α (eIF2α) that leads to the inhibition of IκB translation and the release of NF-κB transcription factors. 45
The fate of cells subjected to DNA damage varies depending on the extent of the damage and the genetic background of the cells. The general paradigm stipulates that while the immediate response to DNA damage, and in particular DSBs, is rapid cell cycle arrest mainly based on the activation of p53, the capacity of the cell to repair the damage determines the rate of recovery and return to the normal cell cycle. 8 If the damage is irreparable or incorrectly resolved, cells stop proliferating definitely and are directed towards apoptosis or senescence.16,46 NF-κB activity is usually described as a prosurvival factor driving resistance to anticancer drug therapies and thus fits well as a factor to promote cell survival during the time needed for DNA reparation. 47 However, sustained NF-κB activation in response to DNA damage then appears contradictory to DDR-induced cell death. In a recent paper, Biton and Ashkenazi 13 showed that the activation of NF-κB by genotoxic agents actually fits this 2-stage cellular response, and they proposed that this pathway may control a switch between a prosurvival and an apoptotic cellular response to DNA damage. In this model, mild DNA damage could promote an early-phase NF-κB response that would be responsible for cell survival during the time required to repair the injuries. In the case of extensive DNA damage, the initial NF-κB response would be maintained and reinforced by a TNF-α/TNFR-1 autocrine loop that is proposed to be responsible for the sustained activation of JNK3. In this model, NF-κB signaling therefore appears to be a fundamental promoter of DNA damage–induced apoptosis. In the next part, we will explore published data that may explain the role of the JNK signaling pathway in apoptosis.
Other Means of JNK Activation by DNA Damage
Since the initial description of JNK activation by DNA-damaging agents, few studies have deciphered the underlying mechanisms involved. Besides the recent breakthroughs described above implicating DNA damage–induced NF-κB activation as an upstream signal of JNK activation, other models explaining the initial observations made on UV-stimulated cells are scarce. The earliest explanation came from Westwick and colleagues, 48 who suggested that ceramides could be activators of the stress kinases. Ceramides are lipid molecules present in the cell plasma membrane that have diverse functions in cellular signaling. 49 Various cellular stresses have been proposed to modulate ceramide metabolism. Among them, DNA damage was shown to induce an augmentation of cellular ceramide levels that are responsible for the activation of JNK and NF-κB signaling pathways.50,51 More recently, Shim and colleagues 52 have shown that TGF-β–mediated JNK and NF-κB activation are impaired in TAK1 knockout cells. 53 This may imply that TAK1’s direct activation by NEMO after DNA damage directly drives JNK activation independently of TNFR. This complements Biton and Ashkenazi’s 13 model stipulating that an autocrine feed-forward loop of TNF-α is responsible for the activation of JNK3 after genotoxic stress. 54
JNK’s activation, as well as other MAPK activations, occurs through a double phosphorylation of a tyrosine and threonine residue in the consensus Thr-X-Tyr (where X is Glu, Gly, or Pro). This activation is negatively controlled by dual-specificity phosphatases that directly dephosphorylate the consensus motif to inactivate the kinase.55,56 Among these phosphatases, MAPK phosphatase 1 (MKP-1) is a cytoplasmic protein that was shown to be down-regulated in cells subjected to translation inhibition induced by severe DNA damage.57,58 The inhibition of MKP-1 translation therefore releases the negative control of JNK and is proposed to drive JNK activation in these conditions.
Cellular Outcomes of JNK Activation by DNA Damage
A wide variety of events occurring downstream of JNK activation, ranging from cytoskeleton remodeling to the regulation of cell proliferation or apoptosis, have been described. In this section, we will focus on JNK activities that may be relevant in the field of DDR, particularly in the control of apoptosis, DNA damage repair, and senescence.
As stated previously, H2AX phosphorylation is one of the most upstream events occurring after a DSB. ATM is the kinase usually described to be responsible for this phosphorylation; however, Lu et al. 59 described that the phosphorylation of H2AX on serine S139 can also be achieved in vivo by JNK. Using either chemical inhibitors, a dominant-negative form of JNKs, and a Jnk2 knockout cell line silenced for JNK1 by siRNAs, they showed that H2AX phosphorylation in response to UV exposure is disrupted only in cells in which both JNK1 and JNK2 activities are hampered. The authors concluded that γH2AX formation is therefore redundantly dependent on JNK1 and JNK2 activities. 59 Moreover, they confirmed that JNK is necessary for caspase-3–mediated apoptosis in response to UV exposure.59-61 The authors proposed a model whereby JNK translocation into the nucleus, where it meets its H2AX substrate, is a core event in the process, leading to UV-induced cell death. Moreover, Lawan and colleagues 62 have shown that UV-induced H2AX phosphorylation and apoptosis are increased in mouse embryonic fibroblasts (MEFs) deficient for the dual-specificity phosphatase MKP-2. Interestingly, MKP-2 is localized specifically in the nucleus, pinpointing the suggestion that specific nuclear activity of JNK is central in JNK-induced apoptosis. 57 Recently, another study showed that the phosphorylation of H2AX by JNK is a very early event in DNA damage–induced apoptosis. 63 Consistent with the idea that H2AX is a substrate for JNK and that γH2AX is required for apoptosis, the authors also described the JNK-dependent pannuclear phosphorylation of H2AX that is proposed to promote the apoptosis of cells subjected to extensive DNA damage. 63 The Fas receptor (Fas)/Fas ligand (Fas-L) is a potent system that induces apoptosis. When Fas-L binds to its receptor, it recruits pro–caspase-8 units that are cleaved into active caspase-8 forms, which in turn activate the caspase-3–dependent cell death pathway. 64 Various genotoxic agents activate Fas-L expression through the activation of JNK and its prototypical targets of the AP-1 transcription factor family.65-68 Apoptosis of cells subjected to genomic insults can therefore also be mediated by a JNK/AP-1/Fas-L axis. Notably, the events leading to DNA damage–induced Fas-L expression are proposed to occur after a prolonged activation of JNK 69 . This raised the question of the role of a potential short-term JNK activity, which is perhaps involved in promoting DNA repair early on after DNA damage is detected.69,70 An interesting observation was made in the late 1990s in which cells deficient for the JNK prototypical target c-Fos transcription factor 71 were shown to be more sensitive to UV-C radiation–induced apoptosis, and this sensitivity was due to an inefficient DNA repair activity. 72 These results are supported by other reports involving members of the Fos-Jun family in DNA repair.73,74 A “ChIP-on-chip” study revealed the promoters binding to c-Jun and ATF2 shortly after the exposure of cells to a genotoxic cisplatin treatment. 75 This study showed that genes encoding DNA repair factors are seemingly predominant among the genes up-regulated by c-Jun and ATF2 immediately after genomic insults. 74 It is noteworthy that γH2AX is proposed to serve as a docking site for DNA repair machinery and is necessary for DNA repair and cell survival after DNA damage.75,76 JNK activity could therefore contribute to the activation of DNA damage repair factors through the phosphorylation of H2AX on S139 in an early phase after genomic insults. Conversely, sustained JNK activation in cells lacking DNA repair is thought to result in apoptosis.
Besides apoptosis, the lack of DNA damage repair can also lead to cellular senescence. Such permanent arrest of the cell cycle is accompanied by various phenotypes including the expression of β-galactosidase, modifications of the secretome of the cells to a so-called senescence-associated secretory phenotype (SASP), and the maintenance of DNA damage foci in the nucleus termed DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS).77-81 While JNK activity has never been implicated in the formation of DNA-SCARS or the expression of β-galactosidase, it is interesting to note that SASP is considered as an inflammatory-like phenotype and that some cytokines belonging to SASP such as IL-1α, IL-6, or IL-8 overlap with the JNK/AP-1–driven inflammatory secretory phenotype.82-86 Moreover, JNK’s homolog p38 MAPK was shown to be a potent inducer of SASP in normal human fibroblasts, mainly through its capacity to enhance NF-κB transcriptional activity. 87 Surprisingly, while JNK’s role in inflammatory phenotypes was described in a variety of cellular contexts, 88 there is still no report of JNK activity being involved in SASP induction. In support of this hypothesis, a recent study demonstrated that induction of the JNK inhibitory phosphatase MKP-1 by gluco-corticoids can counterbalance deleterious effects of JNK signaling in mice. 89 Glucocorticoids are steroid hormones that signal through the glucocorticoid receptor (GR) and display potent anti-inflammatory effects. Using both GR signaling–deficient mice (GR dim/dim mice) and Mkp1–/– mice, Vandevyver and colleagues 89 showed that endogenous glucocorticoids protect the mice from TNF lethal inflammation by up-regulating MKP-1 expression. Moreover, while Jnk1–/– mice displayed no differences in TNF-induced lethality compared to control mice, the Jnk2–/– mice showed better resistance to acute inflammatory stress. The authors concluded that GR signaling counteracts TNF/JNK2-induced inflammation through the up-regulation of MKP-1. 89 Another engaging study has nicely echoed this report, linking glucocorticoids to SASP induction in human cells. 86 In a screen designed to check for compounds able to repress SASP induction in normal human fibroblasts subjected to X-ray irradiation, Laberge and colleagues 86 have shown that the glucocorticoids, corticosterone and cortisol, were able to decrease the secretion of some components of SASP. Notably, both of these glucocorticoids were able to decrease the production of proinflammatory cytokines such as IL-1α, IL-6, and IL-8 through their interaction with the GR. 86 Although the authors proposed that GR-mediated NF-κB inhibition is the key mechanism responsible for the decreased secretion of inflammatory cytokines in glucocorticoid-treated DNA- damaged cells, the potential involvement of JNK in these events is an attractive hypothesis that may deserve future exploration.
The activity of the transcription factor p53, a master regulator of cell fate after DNA damage, has also been linked to JNK signaling. p53 is mainly described to play a pleiotropic tumor suppressor role, as its activation by posttranslational modifications can promote DNA repair, cell cycle arrest, apoptosis, and senescence.70, 90-93 Although over a dozen phosphorylation sites have been mapped in the p53 protein sequence, the phosphorylation of serine 15 by ATM is the best described means of activating p53 after DNA damage. 92 S15 phosphorylation promotes p53 activation by disrupting the interaction of p53 with its inhibitor MDM2.94,95 Threonine 81 of the p53 sequence is a substrate for JNK and appears to be phosphorylated in a DNA damage–dependent manner.96-98 This modification was shown to stabilize p53 and enhance its transcriptional activity, and it appeared to be crucial for p53 activity after genomic insult.97-100 A recent exhaustive study utilizing Drosophila and mammalian cell line models showed that p53 expression is able to potentiate JNK activity. 101 In p53-deficient Drosophila wing imaginal discs irradiated with high doses of γ-rays, JNK activation and apoptosis rates were lower than in the controls. Consistent results were found in p53 knockout MEFs in which the mechanism revealed a binding domain for JNK to p53, where the interaction of JNK and p53 avoids JNK dephosphorylation by MKP-5. 101 As JNK’s capacity to phosphorylate c-Jun is preserved even when bound to p53, the authors proposed a model describing that simultaneous activation of JNK and p53 can cooperate to reinforce JNK-induced apoptosis. 101 Conversely, p53 binding to JNK was also described to hamper JNK activation, thus protecting the cells from UV-induced apoptosis in a lung carcinoma cell line. 102 An explanation for the discrepancies between these 2 studies could stem from the fact that the activity of all 3 jnk gene products was considered as a whole unit in these reports. However, JNK1 and JNK2 are known to mediate opposite effects on p53, with JNK1 proposed to negatively regulate p53, while JNK2 acts as a positive regulator of p53. 103
Overall, the results presented here illustrate the complex interactions involving JNKs in the regulation of the cellular response to DNA damage (Figure 1). In particular, no future studies should exclude assessment of the individual proteins arising from the expression of the 3 jnk genes. Moreover, cell type specificities and the existence of a multiphase cellular response with the immediate activation of pathways promoting cell cycle arrest and survival followed by factors responsible for cell cycle arrest, apoptosis, or senescence can lead to a further variety of responses. Transferring these findings to the clinic proves even more difficult, as additional parameters such as the interactions occurring between different cell types intervene in complex systems such as in vivo tumor growth. In the next part, we will examine some results and hypotheses that may prove relevant in linking genomic stress, JNK activity, and cancer.
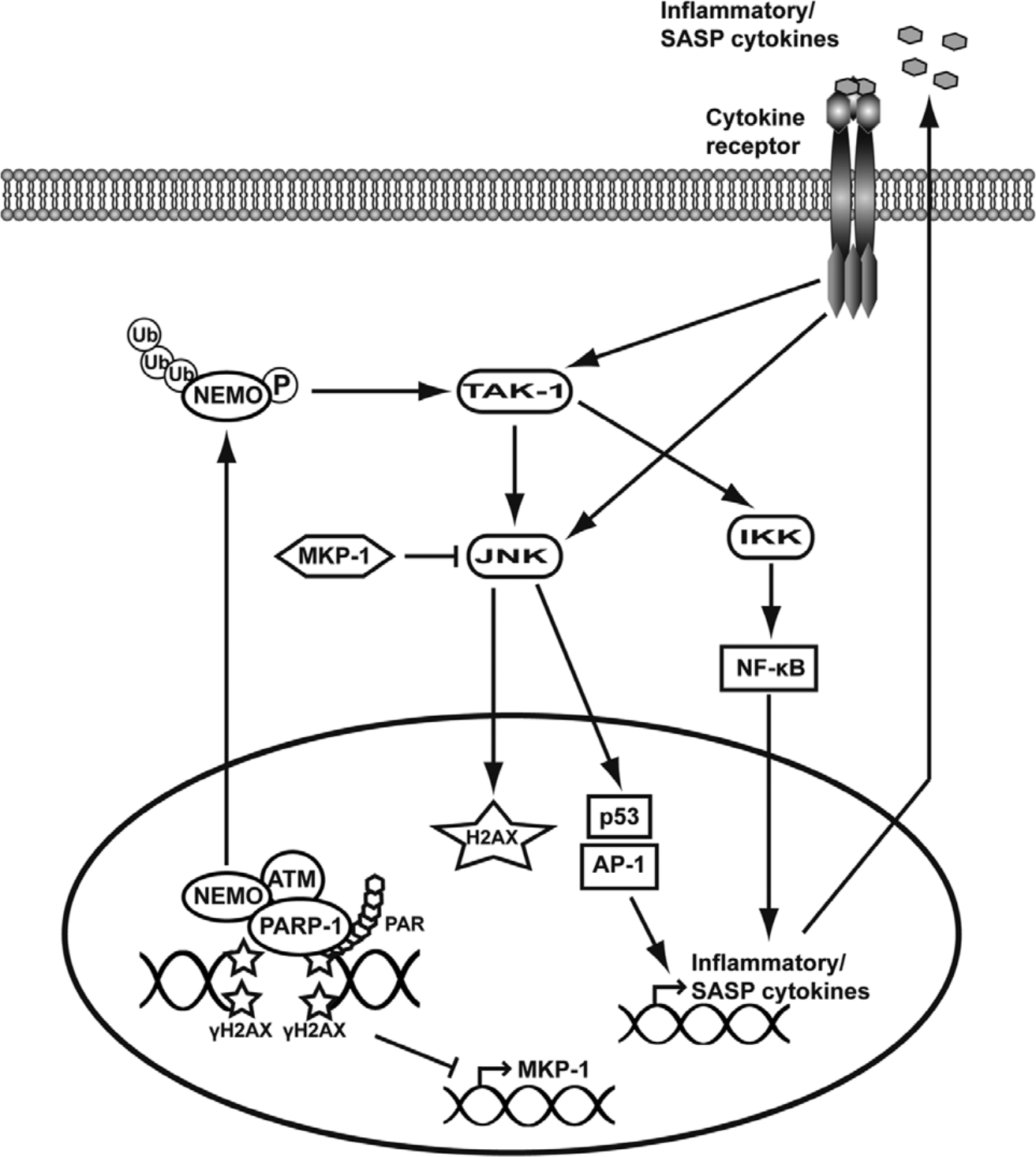
Schematic representation of the pathways proposed to lead to JNK activation following DNA damage. In response to DNA damage, H2AX phosphorylation, recruitment of PARP-1, and subsequent PAR formation at the sites of DNA breakage drive the assembly of a complex comprising at least ATM, NEMO, and PARP-1. This complex is necessary for NEMO posttranslational-activating modifications, such as sumoylation by PIASγ (not represented here), subsequent phosphorylation by ATM, and ubiquitination by cIAP1 (not represented here). Active NEMO is then exported out of the nucleus, where it activates the kinase TAK1, which in turn phosphorylates IKK and JNK directly. Subsequent activation of NF-κB and JNK targets p53, and the transcription factors of the AP-1 family drive the expression of inflammatory cytokines that are proposed to participate to a feed-forward loop responsible for sustained JNK activation. JNK’s kinase activity on H2AX is also proposed to reinforce a DDR. The general repression of transcription after extensive DNA damage is also proposed to account for the activation of JNK via inhibition of the expression of MKP-1, a phosphatase that negatively regulates JNK.
Potential Implications of DNA Damage–Driven JNK Activation in Cancer
In normal conditions, mice and cells depleted of either one jnk or another do not display massive functional difference, pinpointing redundancies in their functions during development and normal cell growth. However, challenging the same cells or animals with nonphysiological constraints such as tumor-inducing or DNA-damaging agents revealed nonredundant and specific roles for each JNK protein.13,104-106 Most striking are the adverse effects of jnk1 and jnk2 knockouts on skin tumor formation induced by treatment with 12-O-tetradecanoylphorbol-13-acetate (TPA). While jnk2–/– mice are less prone to tumor formation in a TPA skin carcinogenesis protocol, jnk1–/– mice exposed to the same protocol form more tumors than the controls.104,105 However, studies concerning the structure of the skin in these mice have shown that jnk1–/– mice display a thinner epithelial layer than jnk2–/– mice, suggesting that differences in the susceptibility toward TPA treatment may result from structural rather than functional differences in the skin epithelium.107-108 The study of JNK and DNA damage interactions may also lead to interesting discoveries in the field of oncogene-induced tumorigenesis. Indeed, oncogene-induced replicative stress leads to genetic instability and activation of the DDR. Senescence or apoptosis of genetically unstable cells therefore represents a major barrier to the transformation of cells. 109 When this barrier is lost, because of a p53 mutation for example, the resulting genomic instability is proposed to drive the transformation of cells.110,111 While JNK activation has been linked to exposure to a variety of DNA-damaging agents, little is known about the capacity of oncogene-induced replicative stress to activate JNK signaling pathways. A recent paper by Chen and colleagues 106 described the oncosuppressive role of JNK2 in a breast cancer model of mice expressing the polyoma middle T antigen (PyV MT) that spontaneously developed mammary tumors. 112 The PyV MT/jnk2–/– mice developed earlier and more frequent tumors as compared to control mice. Moreover, the combined depletion of jnk2 and PyV MT expression induced higher genomic instability with increased aneuploidy and chromosomal rearrangements. Interestingly, nuclei of the tumor cells also displayed less γH2AX and 53BP1 signaling, suggesting that JNK2 may play a role in counterbalancing PyV MT–induced replicative stress by establishing an appropriate DDR. 106 Consistently, the inhibition of JNK2 was shown to induce chromosome segregation defects, resulting in polyploidy in human cervical carcinoma (HeLa) cells and human small-cell lung carcinoma (Calu-1). 113 The authors of this study proposed that JNK1 functions in promoting apoptosis while JNK2 rather regulates progression in the cell cycle.113-115 One could argue, however, that the possible involvement of JKN2 in DNA repair has not been addressed in these studies and hypothesize that JNK2 may be involved more broadly in the maintenance of genome homeostasis. JNK2 disruption would therefore lead to unresolved DNA damage without cell cycle arrest and finally result in the propagation of genomic instability and cancer promotion. Consistent with this idea, the inhibition of basal JNK activity in established breast cancer cell lines was shown to lead to cell cycle aberrations and endoreduplication. 116 More recently, persistent DNA damage induced either by chronic treatment with a DNA-damaging drug or by destabilization of telomeres was shown to lead to similar phenotypes in p53-deficient cells, with the occurrence of endomitosis and tetraploidization events. 117 Taken together, these results allow us to postulate that the disruption of JNK2 activity could participate in DNA damage–induced genetic instability and oncogenesis.
Another potential link between JNK signaling, DNA damage, and cancer was discovered in a study showing that αV integrin is overexpressed in tissue samples from patients with radioresistant nasopharyngeal carcinoma (NPC). 118 Using a NPC cell line cultivated as spheroids, the authors showed that the cell radioresistance is mediated by αV integrin–induced activation of JNK and that JNK inhibition restores the radiosensitivity of the cells. 118 Therefore, studying the role of JNK activity in response to DNA damage could lead to a better understanding of the processes governing the response of cancer cells to DNA-damaging agents.
Further extensive research is required in order to clearly decipher the links existing between DNA damage, JNK signaling, and cancer. In particular, the role of individual JNK isoforms has to be addressed in cells challenged with DNA damage caused by the exposure to DNA-damaging agents such as γ-irradiation or oncogene-induced genomic instability.
Footnotes
Acknowledgements
The authors thank Dr. Scott Parks for editorial assistance.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Institut National pour la Santé et la Recherche Médicale (INSERM), the Centre National de la Recherche Scientifique (CNRS) and the Centre Scientifique de Monaco, the National Institute of Cancer (INCA), the Association for Cancer Research (ARC), the Fondation de France, the Fondation pour la Recherche Médicale (FRM), the “Association pour la Recherche sur les Tumeurs du Rein (ARTuR)”, and ROCHE France.