
Abstract
While recent studies implicate that signaling through the receptor tyrosine kinase MET protects cancer cells from DNA damage, molecular events linking MET to the DNA damage response machinery are largely unknown. Here, we studied the impact of MET inhibition by the small molecule PHA665752 on cytotoxicity induced by DNA-damaging agents. We demonstrate that PHA665752 reduces clonogenic survival of tumor cells with MET overexpression when combined with ionizing radiation and synergistically cooperates with ionizing radiation or adriamycin to induce apoptosis. In search of mechanisms underlying the observed synergism, we show that PHA665752 alone considerably increases γH2AX levels, indicating the accumulation of double-strand DNA breaks. In addition, PHA665752 treatment results in sustained high levels of γH2AX and phosphorylated ATM postirradiation, strengthening the assumption that MET inhibition attenuates postdamage DNA repair. PHA665752, alone or in combination with irradiation, leads also to a massive increase of γH2AX tyrosine phosphorylation and its subsequent interaction with the proapoptotic kinase JNK1. Finally, MET inhibition reduces activation of ATR, CHK1, and CDC25B and abrogates an associated DNA damage–induced S phase arrest. This indicates that MET inhibition compromises a critical damage-dependent checkpoint that may enable DNA-damaged cells to exit cell cycle arrest before repair is completed.
Keywords
Introduction
The currently accepted paradigm for the cellular DNA damage response (DDR) machinery is, that it is comprised of multiple, coordinated cell cycle checkpoint and repair pathways, which detect DNA lesions and signal their presence to the many effectors that participate in the DDR to ensure a prompt repair of the damaged DNA. 1-4 In recent years, a growing body of preclinical and clinical data emerged, which point for potential signaling crosstalks between growth factor receptor tyrosine kinase (RTK) systems and the DDR. 5-8 In this context, MET, the RTK for hepatocyte growth factor (HGF), whose oncogenic activity is highly abundant in numerous types of malignant disorders, 9,10 attracts particular interest, as its efficient targeting is expected to result in both suppression of obvious tumor traits such as metastasis, deregulated angiogenesis, and uncontrolled proliferation, and in tumor sensitization to DNA-damaging agents (DDAs) frequently used in clinical oncology.
In that respect, Fan et al. showed that pretreatment of tumor cells by HGF reduces considerably the formation of DNA double-strand breaks (DSBs) following exposure to ionizing radiation (IR) or adriamycin (ADM). 11 To correlate MET aberrant expression with treatment outcome, Aebersold et al. showed that MET overexpression is a negative marker for radiation therapy treatment in patients with oropharyngeal cancer 12 and that the presence of the activating MET mutation Y1253D in oropharyngeal tumor tissue predicts negative results for local tumor control by definite radiotherapy. 13 Subsequent studies reported that MET inhibition, by a decoy receptor or a MET ribozyme, enhances tumor growth control by IR. 10,14
To elucidate the link between MET and specific DDR pathways, which may underlie tumor resistance to DDAs, we have previously reported that mutated MET variants form an aberrant molecular axis that links this receptor to a pathway that consists of tyrosine kinase ABL and the RAD51 recombinase, two effectors of homologous recombination-dependent DNA repair. 15 Despite these findings, most of the molecular events underlying MET-DDR interactions remain largely unknown.
In the present work, we sought to shed more light over the emerging linkage between MET and the DDR using the anti-MET small molecule PHA665752. 16 The results show increased apoptosis and higher levels of DSBs in cells treated with PHA665752 before exposure to IR or ADM. Calculation of combination indexes (CIs) suggests that PHA665752 is cooperating with IR and ADM synergistically. Our data also imply that PHA665752 alone is able to inflict DSBs in a MET-dependent manner and to delay or attenuate DNA damage repair. Moreover, we provide evidence that MET inhibition is followed by increased tyrosine phosphorylation of γH2AX, which has recently emerged as a crucial molecular event that is associated with postdamage apoptosis rather than DNA repair. 17,18 Finally, we show that MET inhibition results in specific targeting of an ATR-CHK1-CDC25B axis with subsequent disruption of a DNA damage–dependent S phase arrest, providing therefore one potential mechanistic explanation for a MET-DDR signaling pathway.
Results
PHA665752 enhances radiosensitivity of GTL-16 in a clonogenic survival assay
Several studies from recent years have suggested that deregulated MET activity may be associated with cellular radioresistance. 12,19,20 Here, we studied the clonogenic survival of GTL-16 human gastric adenocarcinoma cells, which overexpress MET wt, exposed to various combinations of PHA665752 and IR. Radiosensitivity was not affected by combining IR with 20 nM of PHA665752 as compared to IR alone. However, MET inhibitor used in a 40-nM concentration resulted in remarkably lower clonogenic survival (Fig. 1). In particular, survival at 4 Gy (SF4) was reduced from 53.9% ± 1.0% in the control to 39.1% ± 3.0% in 40 nM of PHA665752-treated cells, while SF4 did not change in cells treated with 20 nM of PHA665752 (54.5% ± 1.0%) as compared to control cells.
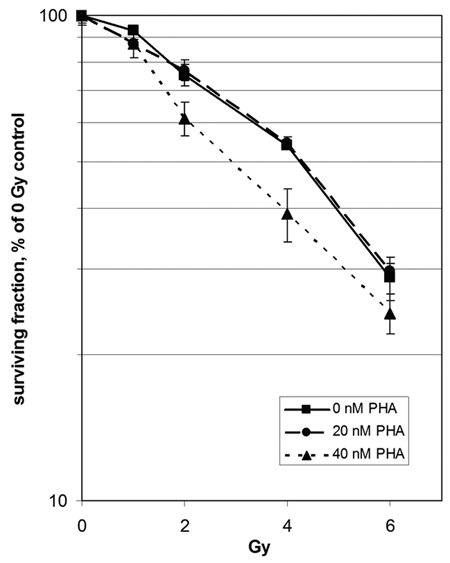
Radiosensitization of GTL-16 cells by PHA665752. For clonogenic assay, cells were pretreated for 24 hours with PHA665752 (PHA) and irradiated. Data points represent the mean surviving fraction ± SEM of 3 independent experiments. Student t test P values combining 40 nM of PHA665752 with 2 Gy and 4 Gy versus without IR are 0.06 and 0.05, respectively.
Inhibition of MET by PHA665752 leads to increased apoptosis upon IR
To investigate if MET inhibition increases IR-induced cell death, we examined the expression of cleaved caspase-3 and nuclear cleaved lamin A in GTL-16 treated by 0, 100, or 300 nM of PHA665752 and subsequently irradiated by 0 to 10 Gy. As Figure 2A shows, the combination of MET inhibition and IR increased the expression of both apoptotic markers 24 hours after IR, while IR alone did not. To confirm these results, we evaluated the impact of PHA665752 used in combination with radiotherapy or chemotherapy on the enzymatic activity of caspase-3. MET inhibition prior to IR increased enzymatic activity of caspase-3 in a concentration-dependent manner (Fig. 2B). Similar results were obtained by combining PHA665752 with the topoisomerase II inhibitor ADM (Fig. 2C), where the use of MET inhibitor significantly increased the activity of caspase-3 in all tested combinations.
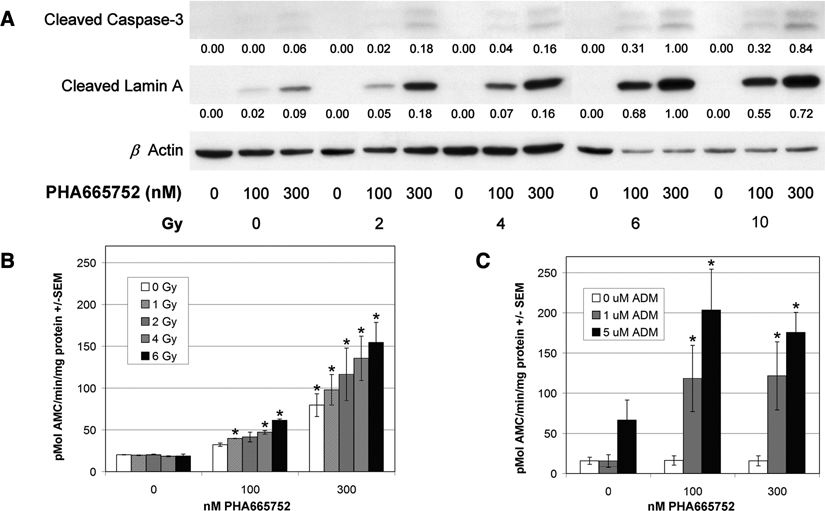
Combination of PHA665752 and IR increases levels of apoptotic markers and enzymatic activity of caspase-3. (
PHA665752 acts with DDAs in a synergistic mode
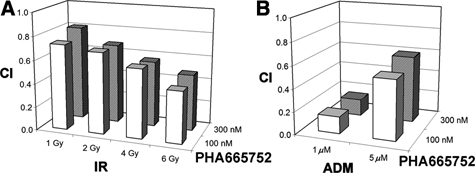
The results described above prompted us to better define the mode by which PHA665752 acts with DDAs to exert increased apoptosis. Using a computational method, 21 we calculated the CIs for PHA665752 and IR or ADM for up-regulating the enzymatic activity of caspase-3 and the expression of cleaved caspase-3 and cleaved lamin A.
For the investigated combined treatments, the CIs related to the effect on caspase-3 enzymatic activity are shown in Figure 3. In all cases, CI values well below one confirmed the synergistic mode of action. Similarly, CIs obtained for the expression of two apoptotic markers show clear synergism between several combinations of PHA665752 and IR (Suppl. Fig. S1).
MET inhibition maintains high levels of γH2AX and pATM after IR
Since compromised DNA repair mechanisms lead to persisting DNA damage, we studied the extent of DSBs in cells treated with IR/ADM alone or combined with MET inhibition by evaluating the levels of Ser139 phosphorylated histone variant H2AX (γH2AX) and of Ser1981 phosphorylated form of ataxia teleangiectasia mutated (pATM), a protein kinase activated by DNA DSBs that generates the initial intracellular DDR signals. 2
Ser139 phosphorylation of H2AX takes place within 1 to 3 minutes after generation of DSBs and is reversed following DNA repair. 22,23 Accordingly, increase in γH2AX and pATM was detected shortly after IR exposure but not 8 hours later (Fig. 4B). Surprisingly, treatment by PHA665752 alone resulted in γH2AX foci (Fig. 4A), indicating that MET inhibition by itself is sufficient to produce DNA DSBs. In the combined protocols, PHA665752 enhanced DNA damage shortly after IR that persisted over a substantially longer time period (Fig. 4B). Likewise, cells treated by ADM exhibited elevated levels of γH2AX, which increased considerably upon combination with PHA665752 (Fig. 4C).
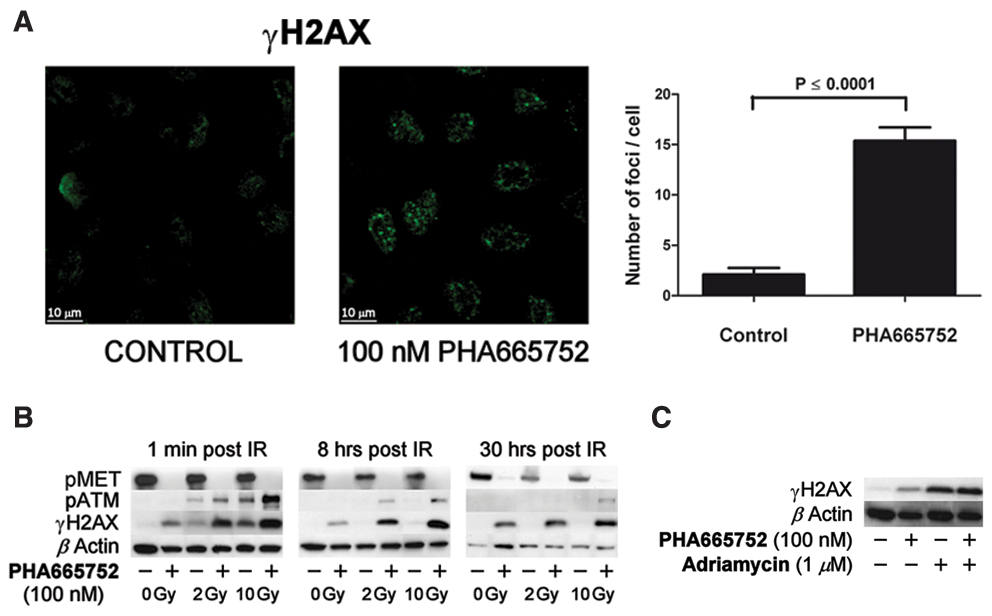
PHA665752 causes DNA damage and suppresses repair of radiation- and ADM-induced DSBs. (
To further support these observations, the effectiveness of MET inhibition to enhance the sensitivity of tumor cells to DNA damage exerted by IR was also shown for the MKN45 and EBC-1 tumor cell lines, which similarly to GTL-16, overexpress the MET receptor (Suppl. Fig. S2). For both MKN45 and EBC-1 lines, a PHA665752 concentration as low as 20 nM was already sufficient to substantially increase γH2AX levels when combined with IR.
DNA-damaging effects of PHA665752 are dependent on MET
To exclude that the PHA665752 effects are due to an off-target activity, we used NIH3T3 cells expressing the MET-mutated variants M1268T and Y1248H that exhibit sensitivity or resistance, respectively, towards another MET small molecule, SU11274. 24 We hypothesized that if MET M1268T and MET Y1248H display a similar distinct pattern towards PHA665752 as towards SU11274, one can use these cells to evaluate downstream effects mediated by PHA665752, which are exclusively dependent on MET. Specifically, we investigated whether MET inhibition by PHA665752 differentially affects cellular DSBs as measured by γH2AX levels in M1268T and Y1248H cells.
As Figure 5 shows, M1268T kinase was responsive to PHA665752 in a dose-dependent manner, reaching total inhibition at 300 nM, while Y1248H autophosphorylation remained unaffected by these concentrations. Consequently, we studied the effect of MET inhibition on DSB formation by evaluating γH2AX status at 1 minute and 8 hours after IR. In the PHA665752-sensitive M1268T cells, exposure to the drug alone elicited DNA damage in a dose-dependent manner, which increased following irradiation, and the administration of 100 and 300 nM of PHA665752 maintained elevated γH2AX levels for 8 hours after IR (Fig. 5A). On the contrary, we could not detect any MET inhibition–dependent DNA damage in the PHA665752-resistant Y1248H cells (Fig. 5B) or in the parental cell line expressing empty vector (Suppl. Fig. S3).
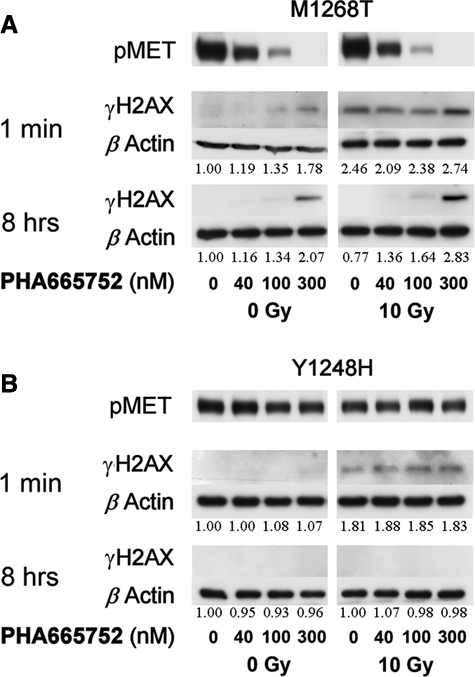
DSBs increase and DNA repair attenuation is mediated through MET inhibition. NIH3T3 cells expressing MET mutations M1268T (
MET inhibition increases γH2AX tyrosine phosphorylation and its subsequent association with JNK1
In two recent Nature articles, Xiao et al. and Cook et al. reported tyrosine 142 as a novel regulatory site of H2AX whose phosphorylation and subsequent dephosphorylation are executed by the WIHC complex and the EYA1/3 phosphatases, respectively. 17,18 H2AX tyrosine phosphorylation serves as a regulatory mechanism, which determines the histone associations with either proapoptotic or repair factors. Overall, persistent tyrosine phosphorylation correlates with γH2AX recruitment of proapoptotic effectors such as the JNK1 kinase, eventually leading to apoptosis. Since γH2AX tyrosine phosphorylation emerges as a novel switch that determines cell fate following DNA damage, we investigated a potential link between MET inhibition and γH2AX tyrosine phosphorylation in irradiated cells. As Figure 6A shows, exposure to PHA665752 was sufficient to considerably increase γH2AX tyrosine phosphorylation even in the absence of DDAs. Interestingly, following a single 10-Gy dose, GTL-16 cells displayed only reduced γH2AX tyrosine phosphorylation, indicating cellular survival. In contrast, cells that were exposed to PHA665752 prior to irradiation exhibited very high levels of tyrosine-phosphorylated γH2AX, reinforcing that MET inhibition compromises cells’ ability to repair DNA damage. To support these observations, we investigated if MET inhibition affects the interaction between γH2AX and the proapoptotic kinase JNK1. MET inhibition alone was sufficient to cause a physical association between γH2AX and JNK1 (Fig. 6B). In accordance with the fact that irradiation was not sufficient to trigger γH2AX tyrosine phosphorylation by itself, γH2AX-JNK1 interaction could not be detected following 10 Gy. However, MET inhibition prior to IR induced a strong interaction between the two proteins.
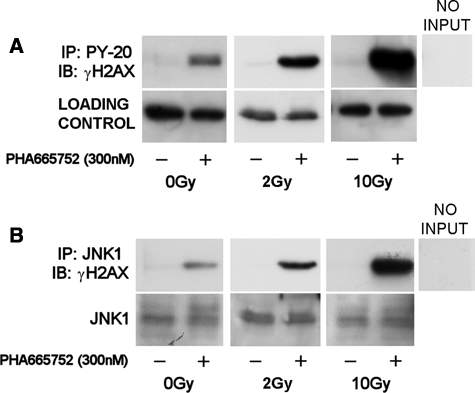
PHA665752 increases γH2AX tyrosine phosphorylation and its association with JNK1. (
MET inhibition targets an ATR-CHK1-CDC25B pathway
As the hitherto data suggest that inhibition of MET activity considerably compromises cells’ response to DDAs, we aimed next at getting an insight into potential MET-DDR signaling pathways. As a preface, it is worthwhile recapitulating that apart from regulating DNA repair, the other major DDR role is to impose molecular checkpoints upon DNA damage. Failure in cell cycle halt is often lethal as it results in detrimental chromosomal aberrations. Targeting this DDR function is therefore considered an attractive direction in current molecular cancer therapy and serves as a conceptual basis for the inhibition of the key checkpoint effectors, kinases CHK1 and CHK2. 25 CHK1/2 reside downstream and are activated by ATM and its related serine/threonine kinase ATR. It is currently accepted that the ATM-CHK2 pathway predominantly regulates the G1 checkpoint, while the ATR-CHK1 pathway controls the S and G2 checkpoints, although there is a crosstalk between these pathways. Checkpoint regulation by CHK1/2 is mediated via phosphorylation of their downstream tyrosine phosphatase, substrates CDC25A/B/C, which is needed to remove inhibitory phosphates from cyclin-dependent kinases for M phase entry. 26 Phosphorylation of CDC25 proteins by CHK1/2 negatively regulates their activity and results in degradation by the proteosome. 26 Here, we investigated a potential link between MET and the ATR-CHK1-CDC25B pathway.
As Figure 7A shows, GTL-16 exhibited basal phosphorylated ATR levels (pATR, Ser428) that marginally reduced following PHA665752 treatment. Exposure of cells to IR resulted in a dramatic increase of pATR levels, which probably reflects attempts to impose checkpoint arrests. This IR-induced pATR increase was however nearly completely abolished in cells where MET was inhibited prior to IR. Since ATR-mediated arrest is transduced primarily via CHK1, we investigated its phosphorylation status (pCHK1, Ser345) at various conditions. GTL-16 displayed moderate basal levels of pCHK1 that were not altered by MET inhibition (Fig. 7A). On the other hand, high pCHK1 levels were detected following irradiation. In accordance with the pATR findings, CHK1 activation appears to be also dependent on MET signaling as its inhibition considerably reduced the phosphorylation of the checkpoint protein. Similarly, irradiation-dependent activation of the potential CHK1 target CDC25B (pCDC25B, Ser187) was reduced when cells were exposed to PHA665752 prior to IR.
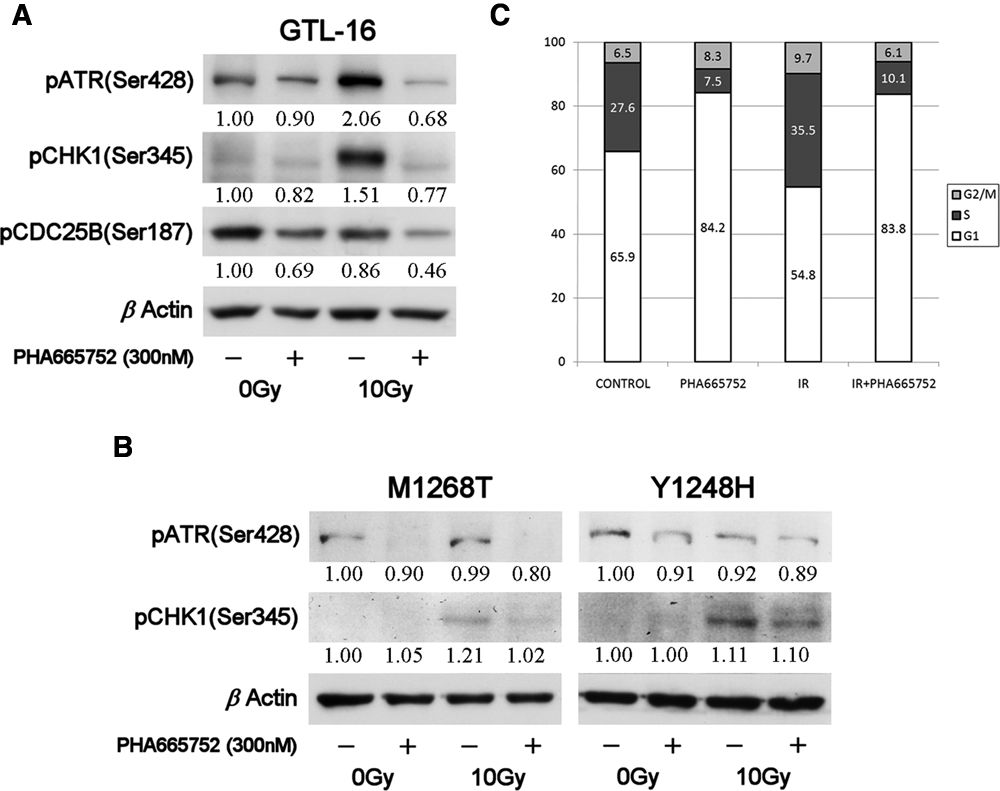
PHA665752 inhibits an ATR-CHK1-CDC25B pathway. (
Subsequently, we questioned whether the observed molecular modulations of DDR effectors by PHA665752 are indeed MET dependent. To that end, ATR and CHK1 phosphorylations have been determined in cells that express the MET M1268T and Y1248H variants. As Figure 7B shows, both basal and postirradiation pATR levels were completely reduced following MET inhibition only in cells expressing the PHA665752-sensitive mutant M1268T, while no considerable impact was detected in the PHA665752-resistant cells Y1248H. Moreover, MET inhibition resulted in a considerable reduction of DNA damage–induced CHK1 phosphorylation in cells expressing the MET PHA665752-sensitive variant, while only a marginal effect was seen in the resistant Y1248H cells.
MET inhibition results in an apparent release from an IR-induced S phase arrest
To verify whether destabilization of a checkpoint that is regulated through the ATR-CHK1-CDC25B axis underlies the MET inhibition–dependent radiosensitizing effects, we tested the impact of PHA665752 on cell cycle distribution in irradiated GTL-16 cells. A single dose of 10 Gy led to an increase of GTL-16 in S phase by nearly 30% as compared to unirradiated controls (Fig. 7C). On the other hand, MET inhibition prior to IR resulted in a considerable drop of S phase cells (35.5% to 10.1%). As CHK1 phosphorylation has been associated with DNA damage–induced intra S phase arrest, our data propose that MET inhibition may eventually release affected cells from growth arrest imposed by this checkpoint, allowing therefore further cell cycle with a load of severely damaged DNA.
Discussion
Although accumulating data suggest that the targeting of RTK systems such as the EGFR, IGF1R, VEGFR, and MET increases tumor cytotoxicity to DDAs, the molecular mechanisms underlying the crosstalk between these receptors and DDR effectors remain largely still unknown. As deregulated MET activity is abundant in human tumors and its link to DNA repair pathways may turn to be a rate-limiting step for treatment outcome when DDAs are used, a better insight into these pathways evolves as an emerging necessity. Accordingly, a prime goal of the current study was to gain an insight into MET-DDR signaling. To that end, we have used PHA665752, a small molecule ATP inhibitor, whose specificity towards the MET kinase activity has been previously documented. 16,27
As PHA665752 increased apoptosis in cells with deregulated MET activity in a synergistic mode when combined with DDAs, we hypothesized that PHA665752 suppresses MET signaling, which is relevant to the repair of DSBs elicited by DDAs. Perhaps one of the most unexpected findings in this set of experiments, summarized in Figure 4, was that MET inhibition by PHA665752 is by itself sufficient to augment γH2AX levels, indicating generation of DSBs. 28 Most probably, the DSBs result primarily by MET inhibition and do not represent late postapoptotic consequences since comparable effects were observed also when cells were treated by PHA665752 only for 2 hours. To our knowledge, this is a first report to suggest that inhibition of an RTK system leads to generation of DSBs. In a previous study, using imatinib mesylate, Liu et al. have reported an increase in γH2AX levels. 29 However, this was noticed only after 72 hours of exposure and with concentrations in the micromolar range. The pattern of γH2AX obtained following the combined treatment protocols provides an explanation for the type of interaction between the MET inhibitor and DDAs. In this sense, it is essential to recall that γH2AX levels seen immediately postirradiation represent the total amount of DSBs, while later time point levels stand for unrepaired DNA. In this respect, even more significant than DSBs, which appear immediately after DDA exposure, are the levels observed at later time points. Any delay in the reduction of γH2AX may result from inhibition of DNA repair. We investigated damage status 8 and 30 hours postirradiation for assessing DNA damage repair. For both time points, considerably high γH2AX levels were maintained in PHA665752-treated cells. Moreover, the results obtained with PHA665752 alone suggest that MET is actively involved not only in the repair of damage caused by exogenous sources but presumably also in the repair of DNA lesions generated under physiological conditions, for example, oxidative stress, which is augmented in highly proliferating tumor cells. 30
Since γH2AX tyrosine phosphorylation has been recently associated with the histone capacity to interact with either apoptosis or DNA repair effectors following DSBs, the observations that MET inhibition triggers γH2AX tyrosine phosphorylation and its subsequent association with the proapoptotic kinase JNK1, even in the absence of IR, provide supportive mechanistic explanations for the aforementioned synergism between PHA665752 and DDAs.
The DDR network executes responses to DNA damage through molecules that function as sensors, transducers, and effectors. 2 H2AX is a key transducing component whose phosphorylation at DSB sites triggers accumulation of other proteins involved in DNA repair and chromatin remodeling. 28 To support the MET-DDR link, we examined the PHA665752 response of the ATM kinase, a major damage sensor located at the apex of the DDR machinery, which is one of the kinases responsible for H2AX phosphorylation. 31 Immediately postirradiation, we detected considerably higher pATM levels in cells with MET inhibition than in cells that were only irradiated, indicating a preconditioning effect for increased DNA damage by MET inhibition. Even more striking was however the fact that at later postirradiation time points, while pATM levels totally declined in cells that were only irradiated, higher levels of this kinase were maintained in cells treated also by MET inhibition. These findings, which parallel those of γH2AX, strongly support the notion that PHA665752 interferes with DSB repair.
Due to its cardinal role in the maintenance of genome integrity, DDR signaling pathways emerge as molecular targets in cancer therapy. 25 Consequently, inhibitors of DDR effectors such as the ATM, Aurora, CHK1/2, and CDK kinases are currently under clinical assessment. 25 In that respect, antagonizing the ATR-CHK1 pathway, a key regulator of S, G2-M, and mitotic spindle checkpoints, is of particular interest. 32 At present, no specific ATR inhibitors have been reported; however, several compounds such as UCN-1, XL844, CHIR-124, AZD7762, and PF-477736, which block CHK1, have been described. 33-37 Inhibiting CHK1 kinase activity is expected to enable DNA-damaged cells to exit cell cycle arrest before repair is completed, leading eventually to a mitotic catastrophe.
As to the current results, our data show that the MET inhibitor PHA665752 effectively compromises the IR-induced DNA damage activation by destabilizing the ATR-CHK1-CDC25B pathway. This is in accordance with previous studies that showed reduction of gemcitabine or irinotecan-induced CHK1 phosphorylation using the CHK1 inhibitors XL844 or CHIR-124. 35,36 Regarding downstream CHK1 signaling, the literature considers CDC25C, and to a lesser extent CDC25A, as the major tyrosine phosphatase substrates of CHK1. 26 Here, we surveyed the impact of PHA665752 on CDC25B, whose biological role is not fully clear yet. Interestingly, our observations that show a consequent reduction of CDC25B phosphorylation in response to CHK1 inhibition by PHA665752 support few previous studies that already suggested CDC25B as a potential CHK1 substrate 38,39 and reinforce the newly described MET-DDR signaling axis. Another important difference between the aforementioned reports that used CHK1 inhibitors and this work is that PHA665752 affects the signaling cascade upstream of CHK1 by blocking already ATR, the major kinase that phosphorylates CHK1. This observation supports our assumption that PHA665752 activity is not elicited through an off-target inhibition of CHK1. This premise was however best validated by the observation that PHA665752 was capable of reducing DNA damage–dependent activation of ATR and CHK1 only in cells expressing the PHA665752-sensitive MET variant, while no parallel inhibitory effects on pATR and pCHK1 were seen in the PHA665752-resistant cells.
Targeting DDR checkpoint effectors such as CHK1 and CHK2 is expected to function through abrogation of their DNA damage–induced cell cycle arrests. 25 For example, depletion of CHK1 by a short hairpin RNA withdraws MDA-MB-231 cells from an S phase arrest induced by the topoisomerase I inhibitor SN38, 40 and similar results were seen when combining gemcitabine with the CHK1 small molecule inhibitor PF477736 in HT29 cells. 33 To validate whether a comparable outcome is seen due to MET inhibition, we examined cell cycle distribution of tumor cells following irradiation. As already mentioned, the data demonstrated a reduction by approximately 70% of S phase IR-arrested GTL-16 cells that were treated by the MET inhibitor in parallel to IR. These findings support the idea that inhibition of a MET-ATR-CHK1-CDC25B axis by PHA665752 leads to abrogation of an S phase arrest, similarly to CHK1 inhibitors.
In summary, the findings presented in this study establish a direct role for an RTK system in the maintenance of the genome integrity via interaction with the cellular DDR machinery. On the therapeutic level, the findings reinforce the potential benefit for the integration of MET inhibitors in cancer therapeutics not only for suppressing tumor growth–dependent MET activity but also for improving the response of tumors with aberrant MET signaling to DNA-damaging modalities, widely applied in clinical oncology.
Materials and Methods
Cell lines
GTL-16 cells were provided by the laboratory of Dr. Paolo Comoglio (Torino, Italy). NIH3T3 cells expressing the MET-mutated variants M1268T and Y1248H were from Dr. Laura Schmidt (National Cancer Institute, Frederick, MD).
Chemicals
PHA665752, (3Z)-5-[(2,6-dichlorobenzyl)sulfonyl]-3-[(3,5-dimethyl-4-{[(2R)-2-(pyrrolidin-1-ylmethyl)pyrrolidin-1-yl]carbonyl}-1H-pyrrol-2-yl)methylene]-1,3-dihydro-2H-indol-2-one (Pfizer, La Jolla, CA) was dissolved in DMSO, and adriamycin (7S,9S)-7-[(2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy- 6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracen- 5,12-dion (Pfizer) in 0.9% NaCl.
Clonogenic assay
Single cells were plated, treated with PHA665752, and 24 hours later exposed to IR using a 137Cs irradiator (MDS Nordion, Ottawa, ON, Canada). One day after IR, PHA665752 was removed. Ten days after plating, cells were fixed and stained with 2% crystal violet. Clonogenic survival was determined using Colcount, Charm Enhanced Algorithmus (Oxford Optronics, UK). Colonies of >50 cells were scored. Clonogenic fraction of irradiated cells was normalized to plating efficiency of nonirradiated controls.
Antibodies
Rabbit anticleaved caspase-3 (Asp175), anticleaved lamin A, and antiphospho-MET (Tyr1234/1235), -ATR (Ser428), -CHK1 (Ser345), and -CDC25B (Ser187) antibodies were all purchased from Cell Signaling Technology (Danvers, MA). Mouse antiphospho-histone H2A.X (Ser139) and antiphospho-ATM (Ser1981) were obtained from Upstate Biotechnology Inc. (Lake Placid, NY). Rabbit anti-MET and mouse anti-JNK1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA), mouse anti-phosphotyrosine PY-20 from BD Biosciences (Franklin Lakes, NJ), and rabbit anti-actin antibody from Sigma (Fluka, Buchs, Switzerland).
Western blotting and immunoprecipitation
Cells were lysed, and protein concentration was determined as described previously. 41 Proteins were resolved by SDS-PAGE, transferred onto PVDF membranes, and incubated with antibodies. Secondary antibodies conjugated to horseradish peroxidase were detected by an ECL kit (Amersham Pharmacia Biotech, Little Chalfont, UK). ECL signals were quantified using Quantity One software (Bio-Rad Laboratories Inc., Hercules, CA).
For immunoprecipitations, lysates were incubated with 1 µg of antibodies, and subsequently, µMACS protein G Microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) were added. After calibration, columns were loaded with samples and washed with high salt and low salt buffers. Beads were boiled with sample buffer and immunoprecipitated complexes analyzed by SDS-PAGE.
Caspase-3 assay
Caspase-3 activity was assessed via a fluorogenic assay utilizing the Ac-DEVD-AMC–specific caspase-3 substrate (Calbiochem, La Jolla, CA). Cells were lysed and analyzed for caspase-3 activity in assay buffer (100 mM HEPES, pH 7.5, 1% sucrose, 0.1% CHAPS). After substrate addition, fluorescence was measured with a TECAN Infinite200 plate reader (Männedorf, Switzerland). Caspase-3 activity was normalized to samples’ protein content.
Confocal microscopy
Cells were prepared as described previously, 15 incubated with anti-γH2AX antibody (1:100), labeled with secondary goat antimouse cyanine 2 antibody (1:50; Juro Supply, Calbiochem, Lucerne, Switzerland), and mounted in PBS:glycerol (2:1) containing 170 mg/mL Mowiol 4-88 (Calbiochem). For analysis, a Zeiss LSM 510 Meta (Carl Zeiss, Oberkochen, Germany) was used. Images were processed using IMARIS software (Bitplane AG, Zurich, Switzerland). Positive γH2AX foci per cell were counted (20 cells evaluated per condition).
Synergism calculations
CIs were calculated according to Chou and Talalay. 21 Briefly, if for a given combination, dosages D C1 and D C2 of drugs 1 and 2, the fraction of affected cells is f A and the drugs are mutually exclusive, CI is defined as CI = D S1/D C1 + D S2/D C2, where D S1 and D S2 are the separate dosages of drugs 1 and 2, respectively, needed to achieve the same affected fraction f A. In our case, values f A, D C1, and D C2 were directly measured, while D S1 and D S2 were inferred from individual dose-effect relations of the used drugs. These relations have the form f A/f U = (D/D m) m , where f A and f U are the affected and unaffected fraction, respectively (f A + f U = 1), D is the dosage of an individual drug, and D m and m are parameters characterizing the drug’s effect; they can be estimated by measuring f A for several different dosages D. Finally, for a particular combination of dosages of 2 drugs or of 1 drug and IR, CI < 1 indicates synergism, CI = 1 indicates summation, and CI > 1 indicates antagonism of the 2 treatment modalities.
Cell cycle
Prior to analysis, fixed cells were rehydrated, centrifuged, washed in PBS, and resuspended in propidium iodide (PI) solution (50 µg PI + 40 µg RNAse A / mL). PI incorporation was measured by FACScan (Becton Dickinson AG, Basel, Switzerland) and analyzed using FlowJo software (FlowJo, Ashland, OR).
Statistics
Statistical analysis of data was performed using the Student t test. Differences with P values <0.05 were considered significant.
Footnotes
Acknowledgements
This study was supported by the Werner und Hedy Berger-Janser Stiftung (grant number 3/2003) and Oncosuisse (grant number OCS-1681-02-2005) grants to Y.Z. and D.M.A.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.