
Abstract
The cellular NAD+/NADH level controls Sir2 (silent information regulator 2) deacetylase activity in regulating aging in lower species. Much work has been put forth to identify ways to activate SIRT1, the mammalian ortholog of Sir2. The identification of p53 as a bona fide substrate of SIRT1 deacetylation has linked SIRT1 to a role in tumorigenesis. Here, we review the various SIRT1 endogenous and small molecular activators and inhibitors that regulate p53 acetylation and subsequent activation of p53 tumor suppression activity.
Sirtuin Background
The sirtuin family represents a unique set of enzymes that respond directly to the energy state of the cell in regulating its intrinsic enzymatic functions. This is achieved during a unique reaction in the enzymatic domain, where in the case of deacetylation, nicotinamide is released from NAD+, and the acetyl group of the substrate protein is transferred to the cleaved NAD+, forming O-acetyl-ADP-ribose, or in the case of ADP-ribosyltransferase activity, an ADP-ribose is transferred to a protein. The mammalian orthologs of yeast Sir2 comprise 7 intriguing proteins that function as deacetylases and/or mono-ADP-ribosyltransferases. SIRT1 is most conserved to its ancestral yeast Sir2, in both protein structure and function, and acts solely as a deacetylase, similar to SIRT2, while SIRT4 and SIRT6 do not have strong deacetylase activity. 1 The most well-studied function of sirtuins is its ability to remove acetyl groups from protein substrates.
Acetylation is historically characterized as a modification of histones; thus, deacetylases are a part of the histone deacetylase (HDAC) family of proteins. Sirtuins belong to the class III HDAC family and are capable of deacetylating histones and many nonhistone protein substrates. The deacetylation of nonhistone proteins has proven to be an area of study that has produced many diverse functions relating back to the sirtuin family members. Deacetylation of proteins, involving everything from cell survival to metabolism, has linked sirtuins to almost every facet of cell regulation. 2
Sir2 was originally identified as a silencing protein in yeast, where its enzymatic activity dictates telomerase silencing and repression of rDNA recombination by regulating chromatin structure. 3 These modifications were demonstrated to be critical to the aging phenotype found in yeast and other lower species. Being an NAD+- dependent deacetylase, it was assumed that the metabolic state of the cell may be contributing to this aging phenotype. The effect on aging was thought to be through the mechanism by which Sir2 regulates caloric restriction. NAD+/NADH levels provide an accurate reflection of the energy state of the cell, where high levels of NAD+ represent a low energy state and high levels of NADH represent a high energy state. 3 The restriction of calories enhances NAD+ levels and reduces NADH levels, resulting in Sir2 activation. Sir2 is able to extend life in lower species by repairing DNA double-strand breaks by nonhomologous end joining and also through its deacetylation of H3 and H4, resulting in the formation of heterochromatin and inhibition of extrachromosomal rDNA circle formation. 3 The functions of Sir2 in lower species are partially conserved in mammalian SIRT1. This revelation led to the expansion of a wide range of research that focused on the role of Sir2 activation by various reagents. One of these activators is resveratrol, which was able to mimic calorie restriction and was thought to reduce the factors that affect aging through SIRT1 activation.4-6 Multiple model organisms were used to examine the effect of various diets and molecules on aging, which resulted in resveratrol and calorie restriction diets gaining a lot of traction in the popular press. However, recently, resveratrol has been demonstrated to not directly interacting with SIRT1, suggesting that the effects of resveratrol may be regulated through another mechanism.7,8 Moreover, much evidence has shown that calorie restriction activation of SIRT1 is conserved in mammalian cells, but the effect is tissue specific. 9 It seems that in a complex cellular environment, NAD+ level is not the only rate-determining factor, as the transcript level of SIRT1 is actively regulated. 10 It was also suggested that SIRT1 expression is regulated by both FOXO3a and mitochondrial biogenesis after changes in the metabolic state of cells, making SIRT1 regulation highly complex in mammalian systems.11,12
SIRT1 has been comprehensively studied in higher organisms, and the focus has been on the substrates of SIRT1 deacetylation. Besides the ability of SIRT1 to deacetylate H1, H3, and H4,13-16 SIRT1 was also found to deacetylate a critical master regulator in p53, suggesting a role in tumor suppression.17,18 Since the initial discovery of SIRT1 deacetylation of p53, many diverse protein substrates have been identified, including FOXO3a, PGC-1α, and PPARγ.19-21 These SIRT1 substrates have linked the metabolic state of the cell to various diseases, including cancer, diabetes, and neurodegenerative diseases.
SIRT1 is biochemically characterized as a 747–amino acid protein that contains 2 pairs of nuclear localization and nuclear export sequences that allow SIRT1 to shuttle between the cytosol and nucleus. Although it has been shown that SIRT1 functions mainly in the nucleus, the ability of SIRT1 to relocalize to compartments in the cytosol allows for potentially cell type–specific functions.22,23 As mentioned earlier, p53 is a deacetylation target of SIRT1; p53 acetylation promotes the transactivation of many specific genes, regulating cell cycle arrest, apoptosis, and metabolic targets. This enzymatic reaction is a key component of p53 regulation of tumor suppression.
p53 Acetylation and Tumor Suppression
Acetylation of p53 and its role in regulating tumor suppression have been extensively investigated in the past 15 years. p53 was the first nonhistone protein found to be acetylated 24 in which acetylation competes to modify lysine residues that are also ubiquitinated, sumoylated, and methylated. 25 This tight regulation of p53 function allows for p53 to react appropriately to different cellular contexts. For instance, when C-terminal lysines are not acetylated, Mdm2 ubiquitinates p53 and targets it for degradation, thus suppressing p53 function. 26 However, when p53 is deubiquitinated by HAUSP, p53 can be acetylated at 6 C-terminal lysines (K370, K372, K373, K381, K382, and K386) by CBP/p300, leading to the transactivation of various p53 transcriptional targets. 27
Recently, novel p53 acetylation sites have been identified that regulate the specific transactivation of downstream targets. PCAF was found to acetylate K320 after genotoxic stress 28 and induce p21-luc transactivation. 29 Acetyltransferases Tip60 and MOF were both found to acetylate K120 on p53.30,31 Acetylation of K120 promoted the transactivation of Puma, leading to apoptosis and sparing cell cycle arrest. Moreover, another acetylation site, K164, was found to be acetylated by CBP/p300. 32 When K164, K120, and the 6 C-terminal lysines were mutated, p21 and Puma transactivations were attenuated. Interestingly, when p53 K120 and K164 mouse homolog sites were mutated, the loss of Puma and p21 transactivations, along with the absence of cell cycle arrest, senescence, and apoptosis, did not lead to tumor formation. 33 Glucose uptake and glycolysis seem to be dysfunctional in these mice, suggesting that other targets that regulate cellular metabolism may be more critical to tumor formation and regulate p53 endogenous tumor suppression activity.
Two groups reported that SIRT1 co-precipitates with p53 and also deacetylates p53 C-terminal lysines, affecting p53 transcriptional activity.17,18 SIRT1 overexpression promoted the survival of cells in the presence of cellular stressors. This molecular mechanism has been verified in many cellular and in vivo contexts. For instance, in one SIRT1 knockout model, thymocytes and fibroblasts, derived from early development, all had enhanced p53 C-terminal acetylation. 34 The SIRT1-deficient thymocytes had enhanced apoptosis after irradiation, suggesting that p53 is activated through acetylation. Another mouse model, MX-cre–dependent, IFN-induced SIRT1 expression in a p53 heterozygous background, had delayed tumor formation and prolonged survival. 35 These mouse models clearly demonstrate the tight regulation of SIRT1 mediation of p53 deacetylation and regulation of p53 tumor suppression activity.
SIRT1 Endogenous Regulators
The significance of SIRT1 deacetylation on p53, a tumor suppressor that is mutated in a large number of tumors, suggests that the SIRT1-p53 axis is a targetable pathway to regulate p53 tumor suppression activity (Figure 1). Over the years, many endogenous factors and small molecules have been identified that can directly affect the SIRT1-p53 pathway. The first endogenous protein that was identified to activate SIRT1 was the active regulator of SIRT1 (AROS). 36 AROS was found to potentiate SIRT1-mediated p53 deacetylation at K382 and inhibited p53 transactivation of downstream effectors p21 and Bax. Knockdown of AROS expression potentiated p53-mediated cell cycle arrest and apoptosis after DNA damage, demonstrating that AROS is an endogenous regulator of the SIRT1-p53 pathway.
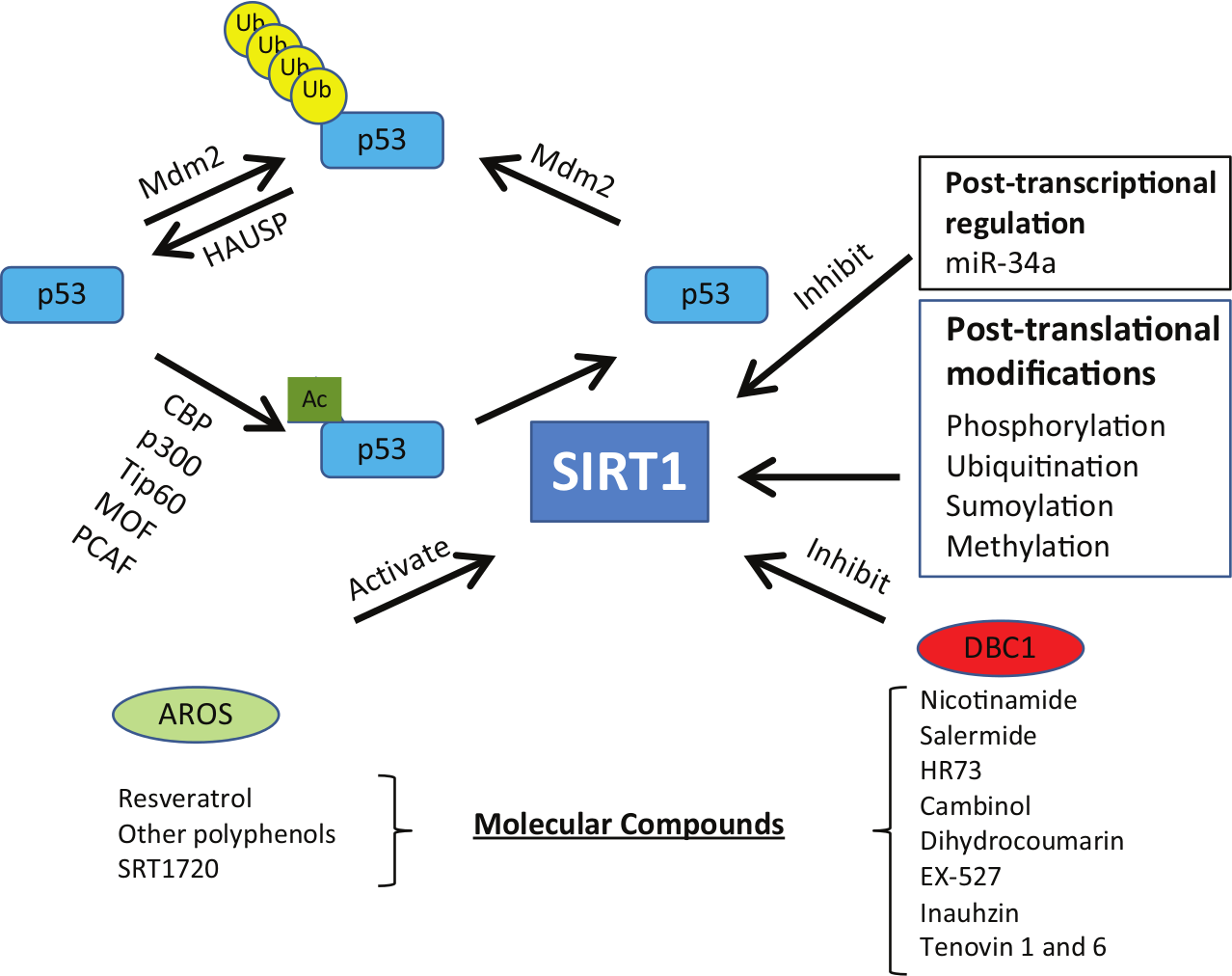
SIRT1-p53 axis regulators.
On the other hand, deleted in breast cancer 1 (DBC1) was discovered to be a strong endogenous inhibitor of SIRT1 activity.37,38 DBC1 interaction with SIRT1 physically inhibited the interaction between SIRT1 and p53, while c-Myc was found to sequester DBC1 from SIRT1, promoting SIRT1 activity in deacetylating p53 K382. 39 The knockdown of DBC1 expression after DNA damage resulted in the attenuation of Puma- and Bax-mediated apoptosis. 38 This response was ameliorated in the absence of SIRT1, demonstrating the necessity of SIRT1 in deacetylating p53. DBC1 knockdown also attenuated FOXO downstream targets, MnSOD and GADD45α expressions, further demonstrating the potency of DBC1 in inhibiting global SIRT1 activity. 37
At the posttranscriptional level, miR-34a was found to inhibit the translation of SIRT1 endogenously. 40 This inhibition led to the enhancement of p53 K382 acetylation and subsequent p53- mediated apoptosis. miR-34a was found to be downregulated in many breast cancer cell lines and 50% of breast cancer tissue samples. 41 In all cases, SIRT1 levels were upregulated as a result of the low levels of miR-34a, suggesting that the absence of miR-34a led to the spike in SIRT1 expression, and this may affect p53 acetylation levels, leading to breast cancer proliferation.
Posttranslational modifications of SIRT1 also have differential effects on SIRT1 function and localization. 42 Mass spectrometry of overexpressed SIRT1 revealed that SIRT1 is an abundantly phosphorylated protein in vivo. 43 Various kinases have been found to regulate SIRT1 activity in a site-specific manner. Casein kinase 2 (CK2) was reported, in vitro, to phosphorylate S659 and S661, 44 while in vivo, after ionizing radiation, CK2 phosphorylated S154, S649, S651, and S689. 45 CK2-mediated SIRT1 phosphorylation increased SIRT1-mediated deacetylation of p53 K382, leading to a decrease in apoptosis after genotoxic stress. More recently, CK2 inhibitors were used to treat glioblastoma cells and efficiently activated p53 function. 46 The cells underwent p53-dependent TNFα-induced apoptosis and also had decreased telomerase activity and induced senescence. CK2 may be a good target for drug development in the hopes of activating p53 tumor suppression activity in existing tumor models. Another kinase that regulates SIRT1 function is c-Jun N-terminal kinase 1 (JNK1). Phosphorylated JNK1 interacts with SIRT1 and phosphorylates SIRT1 at S27, S47, and T530. 47 Phosphorylation of SIRT1 relocalizes the cytosolic population to the nucleus, suggesting that the phosphorylated residues may regulate SIRT1 localization. JNK1-mediated SIRT1 phosphorylation increased the deacetylation of H3 but not p53, demonstrating a specificity in function.
Lysine modifications, such as ubiquitination, sumoylation, and methylation, have also contributed to the regulation of endogenous SIRT1 function. Through mass spectrometry, lysines 254, 335, 377, 499, and 523 were found to be highly ubiquitinated. 48 Although the E3 ligase for SIRT1 is still unknown, USP22 was found to deubiquitinate SIRT1. 48 USP22 knockdown reduced SIRT1 stability, suggesting that the form of ubiquitination on SIRT1 is mainly K48-linked polyubiquitination. When the 5 known ubiquitin-conjugated lysine residues were mutated, USP22 knockdown did not affect SIRT1 stability, suggesting that USP22 deubiquitinated 1 of the 5 residues that were identified. In addition, USP22 overexpression, in the presence of SIRT1, reduced p53 acetylation levels, Puma-luc levels, and apoptosis. These data demonstrate the importance of ubiquitination in regulating SIRT1 protein level and function.
A similar modification to ubiquitination, sumoylation, was discovered to also modify SIRT1. 49 Through the identification of the putative sumoylation sequence and subsequent mutation of the potential sumoylation sites, it was determined that K734 was sumoylated. The mutation of K734 to arginine (K734R) diminished SIRT1-mediated p53 deacetylation at K382, suggesting that sumoylation of SIRT1 enhances SIRT1 acetylation activity. 49 Sumoylation had a global effect on SIRT1 activity since H4 was also demonstrated to be enhanced in the presence of a K734R mutant. SENP1 was found to effectively remove the sumoylation from SIRT1, thus inhibiting SIRT1-mediated p53 deacetylation. Desumoylation was found to be an active mechanism in regulating p53-mediated, genotoxic stress–induced cell death. Methylation sites K233, K235, K236, and K238 were also identified on SIRT1 after doxorubicin treatment. 50 These sites were confirmed to be regulated by Set7/9 methyltransferase. Set7/9 binding to SIRT1 did not affect SIRT1 deacetylation activity on p53 K382 residues. However, Set7/9 overexpression significantly disrupted the SIRT1 interaction with p53; thus, the molecular mechanism that regulates the persistent SIRT1 deacetylation activity in the presence of Set7/9 is still unknown.
SIRT1 Small Molecular Compound Regulators
The search for activators of SIRT1, to replicate the beneficial effects of calorie restriction, led to the identification of the first small molecule to activate SIRT1, resveratrol, which has an effect on extending the life span. Resveratrol is a polyphenol that is extracted from red wine and was found to inhibit SIRT1 deacetylation of an acetylated p53 peptide. 5 Interestingly, recent reports show that resveratrol is not capable of inhibiting endogenous, full-length acetylated PGC-1α and acetylated p53 in vitro, suggesting that resveratrol affects SIRT1 activity indirectly in vivo.7,8 Furthermore, a novel molecular mechanism of action was proposed for resveratrol in which resveratrol directly inhibits phosphodiesterases that degrade cAMP. 51 This leads to a signal transduction cascade that eventually activates AMPK, which increases NAD+ levels to activate SIRT1 activity. 52 A reciprocal relationship between SIRT1 and AMPK was suggested to exist, which was demonstrated utilizing a SIRT1-inducible knockout model in which the presence of SIRT1 in vivo allowed for resveratrol to activate AMPK and increase NAD+ levels. 53 Furthermore, it was also reported that PKA and AMPK may be able to disassociate DBC1 from SIRT1 to further promote SIRT1 activity. 54 Conversely, AMPK was also found to directly phosphorylate T344 residues on SIRT1, inhibiting SIRT1 deacetylation of p53 in HepG2 cells and making the resveratrol-AMPK-SIRT1 axis complex, requiring further elucidation. 55 Since the effect of resveratrol on calorie restriction and SIRT1 may not be clean and direct, there have been strong efforts put forth to identify a more potent SIRT1 activator that is dissimilar in structure to resveratrol. This endeavor is actively being pursued in the pharmaceutical industry and is reviewed in a series of articles.56-58
There are already many SIRT1 inhibitors available for research with varying degrees of potency and specificity. The first inhibitor identified was vitamin B3, or nicotinamide, a by-product of sirtuin enzymatic activity. Nicotinamide was first found to inhibit Sir2 directly in yeast, resulting in increased rDNA recombination and a shortened life span. 59 Since nicotinamide is a noncompetitive intermediate inhibitor of sirtuin enzymatic activity, it is nonspecific in preventing enzymatic activities of all NAD+-dependent enzymes. 60 Interestingly, it was recently reported that in chronic lymphocytic leukemia cells, nicotinamide can be combined with etoposide to effectively activate p53 and transactivate downstream targets that regulate cell cycle arrest and apoptosis. 61 With this in mind, researchers sought to develop a more specific SIRT1 inhibitor that may provide a more potent effect on activating p53 and other SIRT1 substrates.
The first sirtuin-specific inhibitors were identified by phenotypic screens of yeast cultures.62,63 These inhibitors that were identified were specific to Sir2 and were not always effective in inhibiting SIRT1, due to the nature of the screen. One example of this was Sirtinol which was incapable of inhibiting SIRT1 deacetylation of p53 in MCF-7 cells. 64 As a consequence, improvements on sirtinol were made, which led to the synthesis of a structurally similar compound salermide. 65 Salermide was more effective than sirtinol in inhibiting SIRT1 activity; however, salermide was still unable to enhance p53 K382 acetylation. It is quite possible that other p53 acetylation sites are deacetylated since Puma, a p53 downstream transactivation target, is upregulated after salermide treatment. Nevertheless, salermide does not have global effects on the p53 acetylation state, suggesting that the salermide effect may be diluted by some other nonspecific effects.
As for splitomicin, another ineffective SIRT1 inhibitor, 2 new compounds were derived based on its structure. HR73 was developed to be more specific to SIRT1 with higher potency. 66 Another structurally similar compound, cambinol, was constructed and was more soluble and could increase the acetylation of p53, although it still had effects on other human orthologs of Sir2. 67 Interestingly, dihydrocoumarin, which was also identified in phenotypic screens, was found to be a specific inhibitor of SIRT1 activity. 68 Dihydrocoumarin treatment was able to induce a dose-dependent increase in acetylated p53 and apoptosis in TK6 cells.
In an effort to avoid the problems of the first generation of inhibitors developed, Elixir Pharmaceuticals (Cambridge, MA) utilized a high-throughput screen against SIRT1 using a fluorometric assay. A novel class of compounds, indoles, was identified that inhibited SIRT1 at IC50 values of 60 to 100 nM. 69 To great surprise, one of those compounds identified, EX-527, was very specific in inhibiting SIRT1 and enhanced the acetylation of p53 in vivo. 70 More recently, Inauhzin, another indole compound identified by computational screening and cell-based assays, was found to have a profound effect on p53 stability. 71 Inauhzin was found to enhance p53 acetylation by inhibiting SIRT1 activity directly. This led to the attenuation of p53 ubiquitination levels and subsequent degradation, thus activating p53-dependent apoptosis. Further studies examining Inauhzin have demonstrated its efficacy in reducing xenograft tumor sizes in a p53-dependent manner. 72 Indoles may prove to be a very potent class of compounds for clinical use in specifically inactivating SIRT1.
Tenovin compounds were identified similarly by a cell-based screen for compounds that can activate p53. 73 Tenovin-1 was found to be able to activate p53 transactivation of p21. Tenovin-6, a more water-soluble form, was also able to delay tumor growth in xenografts. Both tenovin-1 and tenovin-6 were able to effectively block SIRT1-mediated p53 deacetylation. Unfortunately, even with the robust effects on SIRT1, SIRT2 activity was also compromised, making tenovin compounds effective for tumor suppression but not selective for SIRT1 inhibition.
Conclusion/Remarks
The regulators of the SIRT1-p53 axis have been studied extensively over the recent years. The idea that SIRT1 regulation can activate p53 tumor suppression has identified SIRT1 as a viable target for chemotherapeutics. Most recently, in a chronic myelogenous leukemia progenitor model system that exhibited SIRT1 overexpression, a dual therapy of a tyrosine kinase inhibitor, imatinib, and tenovin-6 was found to be very effective in reducing lymph stem cell xenografts. 74 In these cells, p53 is still active and capable of transactivating downstream targets. Moreover, p53 protein levels correlated with SIRT1 levels in lung squamous carcinoma and adenocarcinoma, suggesting that SIRT1 may regulate p53 stability in various forms of tumors. 75
Most of the data that demonstrated various regulators of SIRT1, affecting SIRT1 deacetylase activity on p53, used C-terminal p53 acetylation as a marker for SIRT1-mediated deacetylation. As mentioned earlier, there are many other transactivation target-specific acetylations not in the C-terminal that may also be regulated by SIRT1 deacetylation. These p53 acetylation sites may end up being more important in regulating p53 tumor suppression by regulating the tumor milieu. 33 The effect on various SIRT1 activators and inhibitors on the acetylation level of these non–C-terminal lysines need to be further examined. Moreover, most of the work on SIRT1 regulation lacked supporting mouse models, with most of the findings demonstrated in vitro or in cell culture. More xenografts and genetic mouse models are needed to assess the role that endogenous and small molecular regulators have on the SIRT1-p53 axis.
The ability of SIRT1 to deacetylate p53 would strongly suggest that SIRT1 functions as an oncogene by inhibiting p53 tumor suppression activity; however, there have also been studies that suggest that SIRT1 may function as a tumor suppressor. 76 SIRT1 deacetylates oncogenes, such as β-catenin and c-Myc, which inactivate their oncogenic properties, leading to SIRT1 behaving with tumor suppression functions.77,78 Mutations in p53 are present in a majority of tumors, which inactivate the SIRT1-p53 axis; thus, it is possible that SIRT1 can function as both a tumor suppressor and activator depending on the genetic profile of the specific tumor.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Cancer Institute of the National Institutes of Health (NIH) under awards 2P01 CA080058-12, 5RO1085533, and 5RO1 CA172023 to W. Gu. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. J.T. Lee was supported by NIH cancer biology training grant T32-CA09503.