
Abstract
Cyclin-dependent kinases (Cdks) drive cell cycle progression in all eukaryotes. Yeasts have a single major Cdk that mediates distinct cell cycle transitions via association with different cyclins. The closest homolog in mammals, Cdk1, drives mitosis. Mammals have additional Cdks—Cdk2, Cdk4, and Cdk6—that represent the major Cdks activated during interphase (iCdks). A large body of evidence has accrued that suggests that activation of iCdks dictates progression though interphase. In apparent contradiction, deficiency in each individual iCdk, respectively, in knockout mice proved to be compatible with live birth and in some instances fertility. Moreover, murine embryos could be derived with Cdk1 as the only functional Cdk. Thus, none of the iCdks is strictly essential for mammalian cell cycle progression, raising the possibility that Cdk1 is the dominant regulator in interphase. However, an absence of iCdks has been accompanied by major shifts in cyclin association to Cdk1, suggesting gain in function. After considerable tweaking, a chemical genetic approach has recently been able to examine the impact of acute inhibition of Cdk2 activity without marked distortion of cyclin/Cdk complex formation. The results suggest that, when expressed at its normal levels, Cdk2 performs essential roles in driving human cells into S phase and maintaining genomic stability. These new findings appear to have restored order to the cell cycle field, bringing it full circle to the view that iCdks indeed play important roles. They also underscore the caveat in knockdown and knockout approaches that protein underexpression can significantly perturb a protein interaction network. We discuss the implications of the new synthesis for future cell cycle studies and anti–Cdk-based therapy of cancer and other diseases.
Regulation of the Mammalian Cell Cycle by iCdks
The centrality of cyclin-dependent kinase (Cdk) roles in eukaryotic cell cycle control was foreshadowed by the discovery that yeast homologs Cdc2 in Schizosaccharomyces pombe and Cdc28 in Saccharomyces cerevisiae (Cdk1 in each) control both G1-S phase and G2-M phase transitions. 1 The paradigm emerged that each transition was driven by the association of the kinase catalytic subunit with different cyclin partners, which direct the complexes to distinct substrates. The ability of Cdk proteins from such evolutionarily distant yeast species 2 and humans 3 to complement yeast cells with conditional defects in Cdk activity underscored the conservation and universality of this regulatory paradigm in eukaryotes.
However, metazoans, 4 including mammals, activate related iCdks in interphase.5-8 These include Cdk4 and Cdk6 during G1 phase and Cdk2 near the start of S phase (Fig. 1). Cdk4/6 bind D cyclins in response to growth factor stimulation and initiate the phosphorylation of retinoblastoma proteins (pRbs).9-12 Cdk2 primarily binds cyclins E and A (A2) and contributes to the phosphorylation of pRbs and NPAT, initiation of DNA synthesis, duplication of centrosomes, and repression of previously fired origins of replication, among other events.13-21 Cdk2 activity peaks in late S-G2 phase, where it appears to contribute to the activation of cyclin B/Cdk1 complexes and entry into mitosis.22-24
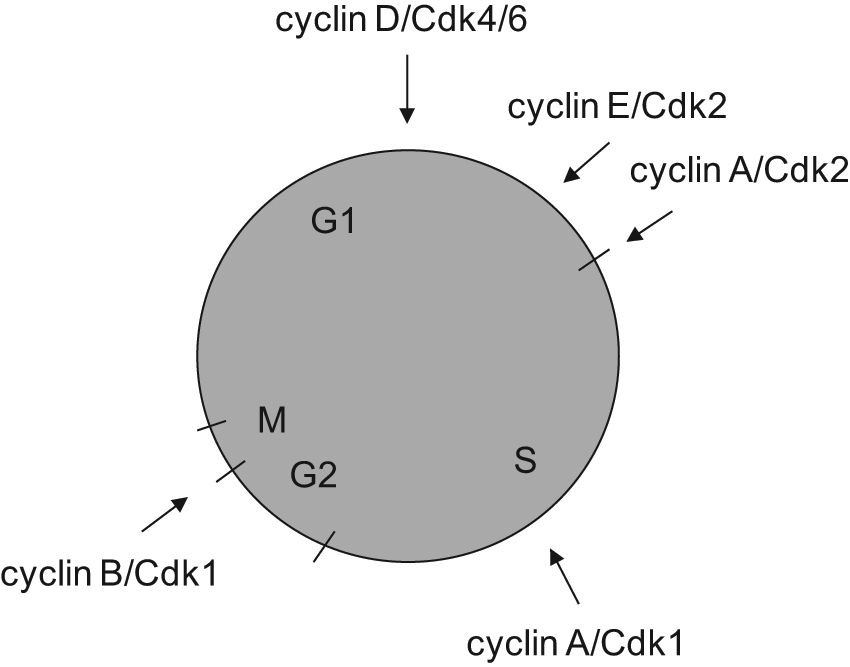
Points of activation of cyclin/Cdk complexes in the mammalian cell cycle.
The evidence that iCdk2, iCdk4, and iCdk6 play important if not essential roles in mammalian cells is impressive for its breadth and depth. iCdks are responsible for most of the Cdk activity leading up to mitosis. Diverse G1 signal transduction pathways activate cyclin D expression and Cdk4/6 activity. 10 Although their net kinase activity is low, their binding affinity and specificity for pRbs are high.12,25-29 Release of pRb-mediated repression of E2F transcription factors drives the induction of cyclins E and then A, followed by a steady increase in overall Cdk activity as cells progress toward mitosis. This increase involves the titration of endogenous Cdk inhibitors from Cdk2 30 complexes as well as their degradation. 31 Microinjection of antibodies directed against the iCdk-associated cyclins D, E, or A 23 each blocked DNA synthesis. Expression of dominant-negative mutants of Cdk4/6 delayed progression into S phase from quiescence, 32 whereas a dominant-negative mutant of Cdk2 was found to block progression into either S phase or mitosis, depending upon the conditions. 22 Native iCdk inhibitor proteins of the Ink4 and CIP1/Kip1 classes were found to be potent cell cycle inhibitors and major or minor tumor suppressor proteins, respectively, without substantial activity toward Cdk1.33-35 Microinjection of cyclin A/Cdk2 complexes into G2 phase cells was found to drive entry into mitosis. 24 Immunodepletion of Cdk2 from Xenopus egg extracts blocked DNA synthesis or, in the absence of DNA synthesis, mitotic entry. 36 Finally, iCdk small molecule inhibitors that harbor little detectable activity against Cdk1 are potent inhibitors of interphase cell cycle progression in vitro and in vivo.37,38 In contrast, Cdk1 activity remains low prior to late S-G2 phase, due primarily to the limited association with cyclins A and B 39 and inhibitory phosphorylation at its ATP binding pocket.40,41 Transient transfection of a dominant-negative mutant of Cdk1 blocked entry into mitosis, 32 and conditional expression reproduces this effect with some late S and G2-M phase delays (G. Enders, unpublished data). A shift of mouse FT210 cells carrying a temperature-sensitive mutant of Cdk1 to the restrictive temperature mediates arrest in G2 phase and perturbed the use of origins of DNA replication in late S phase but does not detectably impact earlier events.32,42-44 These diverse, mutually reinforcing studies lead to a model in which the mammalian cell cycle is initiated by Cdk4/6, S phase is initiated by Cdk2, progression through S and G2 phases is dependent on increasing Cdk2 and Cdk1 activity, and mitosis is initiated by Cdk1 (Fig. 1).
iCdks Are Dispensable
It therefore came as a shock to the field when knockdown experiments suggested that Cdk2 may not be essential in some colorectal cancer cell lines, 45 and knockout mouse studies showed that each of the major iCdks is dispensable for cell cycle progression.46-49 Subtle and/or tissue-specific effects were observed in these constitutive knockout mice. For example, both the Cdk4 and Cdk2 knockout mice exhibited reduced body sizes, and meiosis was defective in Cdk2 knockout germ cells, causing sterility. Cdk4 knockout mice developed insulin-dependent diabetes. Cdk6 knockout mice had reduced thymus and spleen sizes and reduced levels of circulating red blood cells and lymphocytes. Cdk2-null embryo fibroblasts were delayed in moving from quiescence to S phase in vitro, and Cdk2-null neural progenitor cells showed impaired proliferation. These phenotypes might be sufficient to account for the continued conservation of the genes but suggested that iCdks may not be central regulators of the cell cycle. Combination knockout mice generally resulted in embryonic lethality. Nonetheless, embryos lacking all iCdks survived to day 13. Fibroblasts from these embryos cycled more slowly, but they did cycle. In contrast, Cdk1 knockout mice died at embryonic day 2.5 with severe defects in cell proliferation. Clearly, Cdk1 is the only absolutely essential Cdk in the group.
What is going on in mice lacking iCdks? Is cell cycle entry Cdk independent? The answer is clearly no. Indeed, this would have been surprising, given that the major cell cycle transitions are Cdk dependent in all eukaryotes. In iCdk-null cells, S phase now relies on Cdk1, which fills the void. Cdk1 shows an increased association with cyclins D, E, and A, which normally associate more strongly with iCdks. 50 Thus, an absence of iCdks allows the reassortment of cyclins, with enhanced activity of Cdk1 at earlier cell cycle stages.
The more interesting question is whether Cdk1 is in fact the “real” or dominant regulator of the entire cell cycle in normal cells, beyond its widely accepted role as the master regulator of mitosis. The answer to this question also appears to be no. Recall the extensive evidence for roles for iCdks in nonknockout cells and animals. Cdk1 does not appreciably associate with cyclins D, E, and A in most cells until it begins associating with cyclin A in mid-S phase.22,43,51,52 This association pattern reflects the fact that Cdk1 normally loses to iCdks in the competition for binding to cyclins with the exception of cyclin B. The main underlying factor appears to be kinetic differences in activating phosphorylation by the Cdk-activating kinase (CAK)/Cdk7. A series of careful studies from Robert Fisher’s group found that CAK can efficiently phosphorylate the so-called T-loop on monomeric Cdk2, allowing early, stable binding to cyclin A. 39 In contrast, monomeric Cdk1 is a poor substrate for CAK, and complexes between unphosphorylated Cdk1 and cyclin A are unstable. As a result, even though Cdk1 is often expressed at great molar excess to Cdk2, cyclin A/Cdk2 complexes accumulate more rapidly during S phase than cyclin A/Cdk1 complexes.
Chemical Genetic Studies Suggest That Cdk2 Is Normally Essential
Fisher and his group then sought a more definitive test of Cdk2 functions and teamed with Kevan Shokat in a chemical genetic approach. 53 This method, extraordinarily powerful for studying many kinases, including CAK, 39 proved challenging for Cdk2. 43 The method involves making a mutation that enlarges a kinase’s binding site for ATP. The native binding pocket is highly evolved and well defined structurally. The mutant can accommodate binding of the bulky ATP analog inhibitor 3-MB-PP1, thus rendering its kinase activity selectively sensitive to inhibition by this drug (analog-sensitive [AS]). If the mutant otherwise retains sufficient normal activity, in the absence of the drug, it can be substituted for the native kinase by homologous recombination of the gene. Homozygous substitution renders the entire activity of the kinase in the cell sensitive to the drug. The problem with Cdk2 was that the enlarging mutation, a substitution of glycine for the bulky phenylalanine residue at position 80, compromised Cdk2 activity in the absence of the drug. Undeterred, Shokat and Fisher studied this problem and found that the mutant enzyme was deficient in binding to cyclin A. Based on structural considerations, they reasoned that incubation with an ATP analog that carries specific but low affinity for the mutant ATP binding site might stabilize cyclin A binding yet permit exchange with either ATP, for normal activity, or the bulky ATP analog, for potent inhibition. This was exactly what was observed. Incubation of cells with the drug 6-benzylaminopurine restored normal cyclin A binding and allowed activity of the complex in the presence of ATP. Addition of the bulky analog 3-MB-PP1 inhibited activity. 43
Effects on human cell proliferation were tested in both transformed HCT-116 colorectal carcinoma cells and nontransformed hTERT-immortalized retinal pigment epithelial (RPE) cells. Recovery of proliferation following serum starvation (both cell types) or contact inhibition (RPE cells) was inhibited in Cdk2 AS/AS double-knockin cells treated with 3-MB-PP1. Proliferation of control cells lacking AS alleles was unaffected. Time course experiments suggested the inhibition of progression through both the restriction point, the last growth factor–dependent point before S phase, and the subsequent G1-S transition. 43 These roles for Cdk2 do not appear to be unique to human cells. Cdk2-null mouse embryo fibroblasts engineered to overexpress Cdk2 AS were actually more sensitive to inhibition by 3-MB-PP1 than human Cdk2 AS/AS cells, although competition of the overexpressed protein with Cdk1 in cyclin A binding may have contributed to the sensitivity. 52 Follow-up chemical genetic studies revealed a role for Cdk2 in survival following ionizing radiation, 54 confirming and extending earlier evidence for Cdk1/2 roles in the DNA damage response.55-60
Thus, a consensus seems to be emerging that Cdk2 normally initiates DNA synthesis and mediates progression to mid- to late S phase. At this point, Cdk1 also begins to associate substantially with cyclin A. The combined action of cyclin A/Cdk2 and cyclin A/Cdk1 complexes completes S phase and primes the activation of cyclin B/Cdk1 complexes for the initiation of mitosis. 61 Targets of Cdk1 during late S phase include origins of replication, the anaphase-promoting complex subunit Cdh1, and a histone mRNA binding protein.44,51,62
It is less clear whether Cdk4 and Cdk6 are broadly pivotal in driving entry into the cycle, although they appear to be so in select tissues such as hematopoietic cells and pancreatic islets.46,48 These Cdks are much less conserved than Cdk1 and Cdk2, do not rescue functions of yeast Cdk1 mutants, exhibit far less kinase activity, have major noncatalytic roles in the activation of Cdk2, and have a narrow substrate range. Cyclin D/Cdk2 complexes have been found to be catalytically inactive but might play noncatalytic roles in titrating iCdk inhibitor proteins and activating cyclin E and A complexes. There is substantial evidence that cyclin D and E are in part dispensable.63-65 In contrast, a cyclin A (A2) knockout mouse is lethal with marked impairment of stem cell proliferation. 66 Cdk1 and Cdk2 can also phosphorylate pRb proteins, 28 and there is considerable evidence that Cdk2 actually does so in normal cells. 21 The substrate preference for cyclin D/Cdk4/Cdk6 complexes is different than for Cdk2 complexes. 67 Some pRb phosphorylation sites have been shown to be relatively specific for Cdk4/6 complexes, but these have not been shown to be pivotal.68,69 Thus, Cdk4 and Cdk6 are clearly more specialized than Cdk1 and Cdk2 and not as central to the cell cycle. Nonetheless, there is considerable evidence, under conditions of normal expression levels, that Cdk4/6 activity is needed for cell cycle progression in many cells, not least of which is the potency of relatively Cdk4/6-specific drugs. 70
Implications for Future Cell Cycle Studies
The new chemical genetic evidence for important roles for Cdk2 in S phase entry mandates the reassessment of methods used in cell cycle studies. This work underscores that Heisenberg’s uncertainty principle applies even to the gold-standard knockout/knockdown approach to defining a protein’s role. That is, any interrogation of a system perturbs it, in this case via protein underexpression. In a highly conserved protein family such as the Cdks that relies on competitive binding to somewhat promiscuous cyclin co-factors, Cdk2 underexpression allows a substantial reassortment of cyclins to Cdk1, masking normal roles of Cdk2. In retrospect, the ability of Cdk1 to compensate for the absence of Cdk2 should perhaps not be surprising. The primary structure of Cdk1 is slightly more conserved than that of Cdk2, compared to the canonical yeast Cdks. iCdks may have evolved in metazoans by gradually augmenting and usurping roles previously performed by Cdk1. Given the potential for Cdk inhibitor drugs37,38 to produce off-target effects, for dominant-negative mutants to titrate cyclins away from other Cdks, for knockout and knockdown to cause a redistribution of cyclins to other Cdks, and for injected antibodies to have off-target or complex effects, the chemical genetic method appears to be least prone to artifacts, as long as the target kinase can be engineered without crippling its complex formation and activity in the absence of a strong inhibitor. The chemical genetic method therefore offers investigators the potential to confirm and discover kinase roles under fairly physiological conditions and gain greater control over the timing and level of kinase activity. On the other hand, the other methods are simpler and have merit. Moreover, it is unclear as yet whether Cdk AS mutants can function effectively in knockin studies in mice. The current need for Cdk2 AS mutants to be treated with a low-affinity ATP inhibitor to support cyclin binding is an obvious disadvantage for such work.
Implications for Cdk-Based Drugs in Medicine
The broad dysregulation of Cdks in cancer has made them attractive potential targets for cancer chemotherapy.33,38,59,71 For example, cyclin D1 was identified early on as a target of a chromosomal rearrangement in a parathyroid tumor. 72 Cdk4 is amplified in some sarcomas. 73 Cyclin E is amplified and/or overexpressed in some breast tumors.74,75 Although there is substantial evidence for some Cdk-independent activities of cyclins,76-78 they remain less established than their roles in activating Cdks. Recent genomic sequencing studies confirm that cyclins and Cdks are objects of potential oncogenic mutation in several cancers, often by gene amplification.79,80 The knockdown and knockout studies of Cdks45-47,50 remain quite valuable contributions to our understanding. They point out that cancer cells that eliminate the expression of some iCdks may become resistant to iCdk-based drugs while maintaining Cdk function through enhanced cyclin binding to Cdk2 and/or Cdk1. On the other hand, the chemical genetic data, combined with the wealth of evidence obtained by other methods, suggest potential therapeutic roles for iCdk inhibition, particularly in tumors with specific upregulation of iCdk activity. Loss of iCdk expression may not be sustained so easily in this context. Substantial evidence exists that cancer cells can exhibit an “addiction” to the activation of dysregulated signaling pathways. 81 The breadth and potency of tumor suppression by the native iCdk inhibitor p16Ink4a, which includes the suppression of advanced tumors,82,83 imply that iCdk pathways are often crucial.33-35,84,85 Cdk1 may not fully compensate for the loss of an iCdk in some cancer cells. Toxicity in normal cells remains a substantial concern, perhaps more so given the recent evidence that iCdks are indeed needed for normal cell proliferation. Whether a net benefit can be achieved in cancer chemotherapy using iCdk inhibitor drugs remains to be established. Cdks have also been suggested as drug targets in other diseases that involve cell proliferation, such as arthritis, in which the proliferation of inflammatory and immune cells is prominent. 86 In these instances, selection for the loss of iCdks is less of a concern.
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received the following financial support for the research, authorship, and/or publication of this article: Work in the author’s laboratory is supported by National Institutes of Health grant AG040366 (G.H.E.), Fox Chase Cancer Center/National Cancer Institute grant P30 CA006927, and the Epigenetics and Progenitor Cell Keystone and Cancer Biology Programs.