
Abstract
Neuroblastoma (NB), the most frequent extracranial solid tumor of children accounting for nearly 15% of all childhood cancer mortality, displays overexpression of antiapoptotic Bcl-2 and Mcl-1 in aggressive forms of the disease. The clinical phase 2 drug roscovitine (CYC202, seliciclib), a relatively selective inhibitor of cyclin-dependent kinases (CDKs), and CR8, a recently developed and more potent analog, induce concentration-dependent apoptotic cell death of NB cells (average IC50 values: 24.2 µM and 0.4 µM for roscovitine and CR8, respectively). Both roscovitine and CR8 trigger rapid down-regulation of the short-lived survival factor Mcl-1 in the 9 investigated human NB cell lines. This effect was further analyzed in the human SH-SY5Y NB cell line. Down-regulation of Mcl-1 appears to depend on inhibition of CDKs rather than on interaction of roscovitine and CR8 with their secondary targets. CR8 is an adenosine triphosphate-competitive inhibitor of CDK9, and the structure of a CDK9/cyclin T/CR8 complex is described. Mcl-1 down-regulation occurs both at the mRNA and protein levels. This effect can be accounted for by a reduction in Mcl-1 protein synthesis, under stable Mcl-1 degradation conditions. Mcl-1 down-regulation is accompanied by a transient increase in free Noxa, a proapoptotic factor. Mcl-1 down-regulation occurs independently of the presence or up-regulation of p53 and of the MYCN status. Taken together, these results suggest that the clinical drug roscovitine and its novel analog CR8 induce apoptotic tumor cell death by down-regulating Mcl-1, a key survival factor expressed in all NB cell lines. CDK inhibition may thus constitute a new approach to treat refractory high-risk NB.
Introduction
Neuroblastoma (NB), the most frequent extracranial solid tumor of children, derives from immature sympathetic neuronal cells. NB accounts for 10% of all childhood cancers and nearly 15% of all childhood cancer mortality. 1-3 These pediatric solid tumors, which begin in early childhood, show extreme heterogeneity at the clinical, histological, and genetic levels. Among the diversity of genetic variations, MYCN amplification is a genetic hallmark of the disease and an independent marker of dismal prognosis. 4 A panel of prognostic factors, including age at diagnosis, tumor burden, histopathology, DNA index, and MYCN status, is used to determine risk categories. 5 In a recent study, genome analyses have been used for the genomic stratification of all clinical forms of NB at diagnosis. 6 About half of NBs are considered high risk at diagnosis. These consist of MYCN-amplified NB as well as stage 4 NB in patients older than 12 months. High-risk NB (HR-NB) patients are characterized by distant metastases and an aggressive course with bleak prognosis. Numerous studies have been conducted to identify the genes involved in the aggressive forms of this disease, but only about 25 genes, including MYCN, have been proven or are likely to be involved in NB tumorigenesis, invasion, and dissemination. 4,7,8 From a mechanistic viewpoint, HR-NB, albeit harboring wild-type p53, 9 are unable to undergo apoptosis, show a marked defect of caspase 8, 10 and elicit specific survival mechanisms. 11 Most HR-NB patients will have poor prognoses (about 25% overall survival at 5 years) despite intensive high-dose and myeloablative chemotherapy, as well as a rescue therapy involving autologous hematopoietic stem cell transplantation, followed by a maintenance regimen of 13-cis-retinoic acid. 12 As conventional, intensive multimodal therapies are insufficient for HR-NB, 13 there is a strong need for new first-line molecular-based therapeutic approaches.
Among the 518 human kinases, cyclin-dependent kinases (CDKs) have been extensively studied because of their key functions in fundamental cell processes such as regulation of cell division and cell death. 13,14 Numerous abnormalities in the regulation and activity of CDKs have been observed in cancers, and this has encouraged the search for, as well as optimization and detailed characterization of, pharmacological inhibitors of CDKs as potential anticancer agents (reviews in references 14-19). Over 140 CDK inhibitors have been described, and 10 of these are currently undergoing phase 1 or phase 2 clinical investigation. 18,19 Among these, the 2,6,9-trisubstituted purine (R)-roscovitine (CYC202, seliciclib) 20 (reviews in references 21-23) is in late phase 2 trials against non–small cell lung cancer and nasopharyngeal cancer. 24,25 However, its short half-life, its inactivating metabolism, 26-28 and its rather weak potency on CDKs and cell lines and, as a consequence, the large quantities required to treat patients constitute limiting factors in its clinical use. Thus, second-generation analogs of roscovitine, in which improved potency would be combined with the rather good selectivity and limited toxicity of roscovitine, are needed. In this context, we have explored the purine scaffold and its analogs rather extensively. These efforts have led to the identification of the CR8 series, a family of compounds with improved potency at inducing cell death. 29,30 Despite modest improvement in its activity as a CDK inhibitor, (S)-CR8 induces apoptotic cell death with about 50- to 200-fold enhanced potency compared to (R)-roscovitine. Deciphering the molecular mechanism of action of such potent compounds may afford an innovative approach to the treatment of HR-NB, which is well known for being refractory to apoptosis.
Roscovitine and other CDK inhibitors have been shown to trigger rapid down-regulation of the survival factor Mcl-1 (myeloid cell leukemia 1) in chronic lymphocytic leukemia, 31-33 multiple myeloma, 34,35 and nasopharyngeal carcinoma. 36 Mcl-1 is an antiapoptotic Bcl-2 family member that is highly expressed in numerous tumor cells but poorly or not at all expressed in normal cells (reviews in references 37-39). Mcl-1 is a very short-lived protein that is rapidly degraded by an ubiquitin-dependent pathway. Increased levels of the deubiquitinase USP9X and subsequent stabilization of Mcl-1, resulting in abnormally high levels of Mcl-1, strongly contribute to tumor cell survival. 40 Recent observations have shown a correlation between high expression of Mcl-1 and Bcl-2 and HR-NB. 41 RNA interference experiments showed that Mcl-1 knockdown induced apoptosis in NB cell lines and sensitized them to cytotoxic chemotherapy. 41 These results prompted us to investigate the effects of CDK inhibitors on Mcl-1 expression in NB cells. We here report that CDK inhibitors trigger rapid and extensive down-regulation of Mcl-1 expression in NB cells, followed by transient free Noxa up-regulation and cell death. Mcl-1 down-regulation is seen in all NB cell lines investigated, independently of their p53 and MYCN status. Mcl-1 down-regulation by CDK inhibitors may thus constitute a new therapeutic approach to be investigated further in HR-NBs. This is particularly well supported by the recent discovery that inactivation of CDK2 is synthetically lethal to MYCN overexpressing cancer cells. 42
Results
Roscovitine-derived CR8 is a potent inducer of neuroblastoma cell death
(S)-CR8 has been optimized as a second-generation derivative of (R)-roscovitine (Figure 1). 29,30,43 The compounds will be referred to as CR8 and roscovitine throughout this study. Although slightly more potent than roscovitine as an inhibitor of various kinase targets, CR8 was found to be much more potent than its parent molecule at inducing cell death in a variety of cells (average 59-fold higher potency on 6 cell lines, as described in Bettayeb et al., 29 and an average of 102-fold higher potency on the NCI 60 cell line panel [data not shown]). We first compared the effects of CR8 and roscovitine on cell survival of 9 human NB lines, including SH-SY5Y (Table 1). Both compounds induced concentration-dependent cell death in all cell lines (Table 1). CR8 showed an average 60-fold higher potency over roscovitine in the 9 NB cell lines. Synchronization of SH-SY5Y cells with either hydroxyurea or nocodazole did not modify their sensitivity to apoptosis induction by roscovitine and CR8, suggesting that the drugs can induce cell death in any cell cycle phase (data not shown).
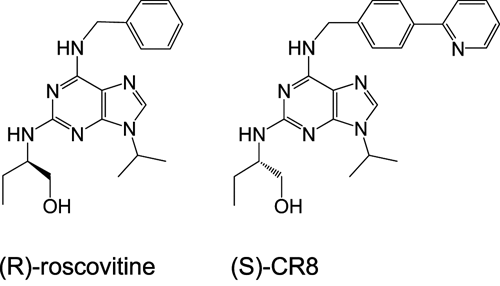
Chemical structure of roscovitine and CR8.
Effects of Roscovitine and CR8 on the Survival of Human NB Cell Lines
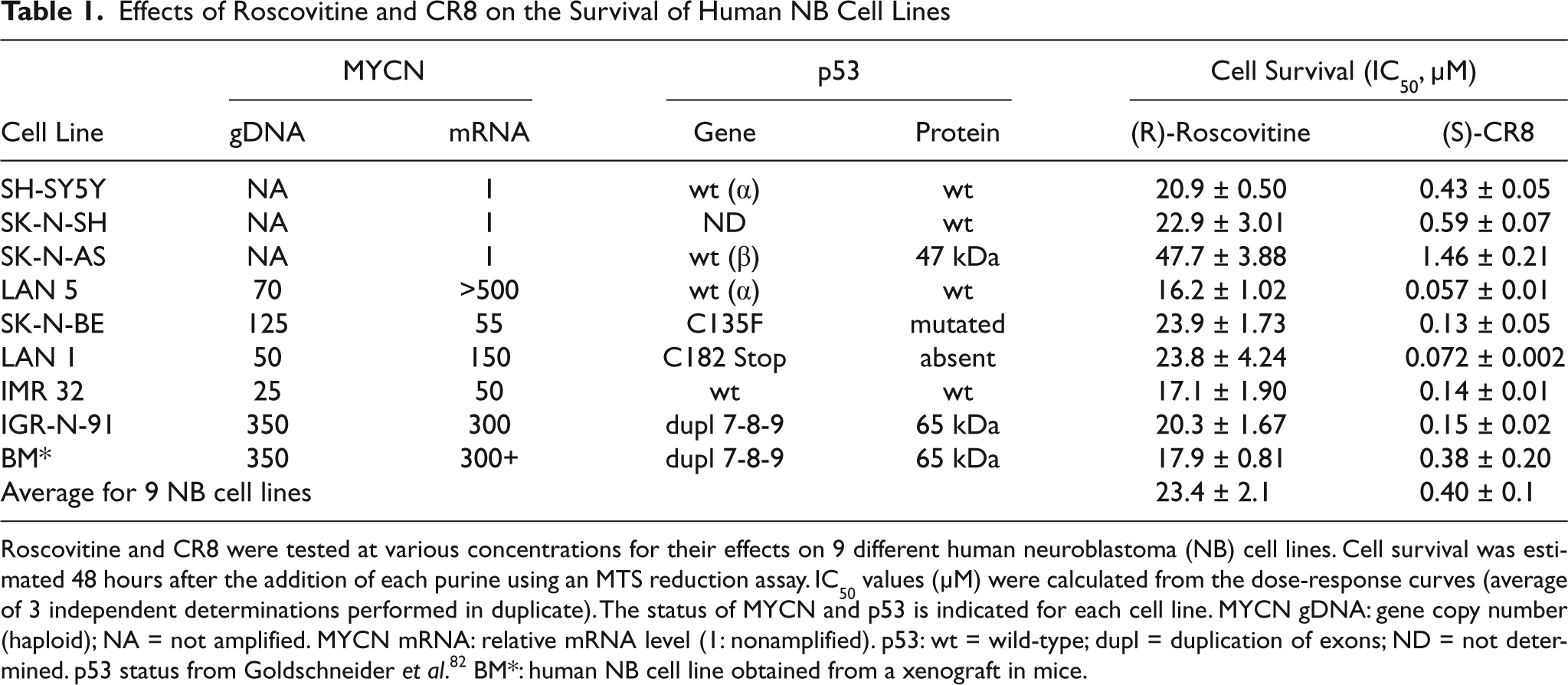
Roscovitine and CR8 were tested at various concentrations for their effects on 9 different human neuroblastoma (NB) cell lines. Cell survival was estimated 48 hours after the addition of each purine using an MTS reduction assay. IC50 values (µM) were calculated from the dose-response curves (average of 3 independent determinations performed in duplicate). The status of MYCN and p53 is indicated for each cell line. MYCN gDNA: gene copy number (haploid); NA = not amplified. MYCN mRNA: relative mRNA level (1: nonamplified). p53: wt = wild-type; dupl = duplication of exons; ND = not determined. p53 status from Goldschneider et al. 82 BM*: human NB cell line obtained from a xenograft in mice.
Roscovitine and CR8 trigger rapid down-regulation of Mcl-1 mRNA and protein
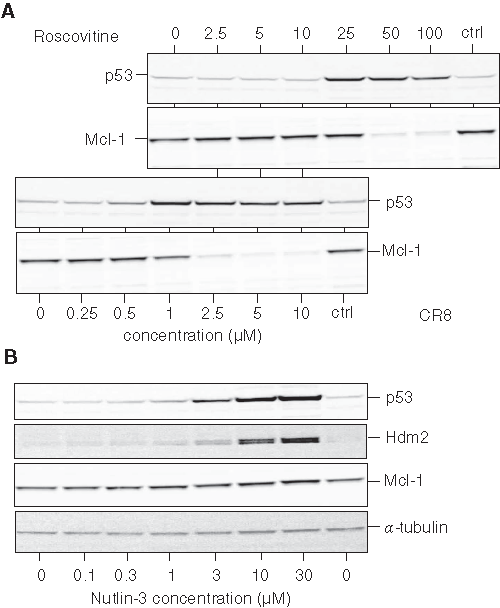
To investigate the mechanism of action of CR8 and roscovitine on NB cells, we selected the human NB SH-SY5Y cell line for further study. Cells were exposed to 5 or 50 µM of each drug for various durations. mRNAs were extracted and reverse-transcribed, and the corresponding Mcl-1 cDNA was amplified with specific primers. Results show that, at 50 µM, both compounds induced a rapid down-regulation of Mcl-1 gene expression (Figure 2). However, at 5 µM (a concentration insufficient for roscovitine to trigger cell death but sufficient for CR8), Mcl-1 gene expression was down-regulated only by CR8.
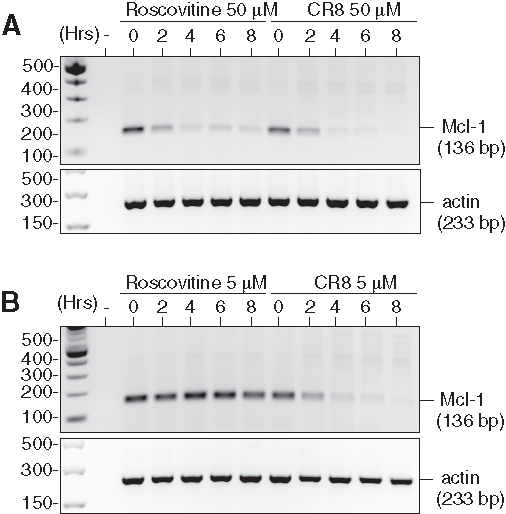
Roscovitine and CR8 induce massive down-regulation of Mcl-1 mRNA. SH-SY5Y NB cells were exposed for various durations to roscovitine or CR8 at either a (
When Mcl-1 protein levels were analyzed under similar conditions, a time- (data not shown) and concentration-dependent (Figure 3A) down-regulation was observed. Complete Mcl-1 down-regulation was observed at much lower concentrations of CR8 than roscovitine and was accompanied by the appearance of low MW Mcl-1 proteolytic fragments, some of which have been reported to display proapoptotic properties. To investigate whether Mcl-1 down-regulation induced by roscovitine and CR8 was general in NB, we tested 8 additional human NB cell lines. In all cases, both drugs triggered a dramatic concentration-dependent decrease in Mcl-1 protein expression (Figure 3B; Suppl. Fig. S1). This suggests that Mcl-1 down-regulation is not restricted to SH-SY5Y and might represent a broad mechanism of action of CDK inhibitors in human malignant NB cells. Essentially, all cell lines responded at similar concentrations, with the exception of SK-N-AS and SK-N-SH, which were slightly less sensitive to CR8 and roscovitine.
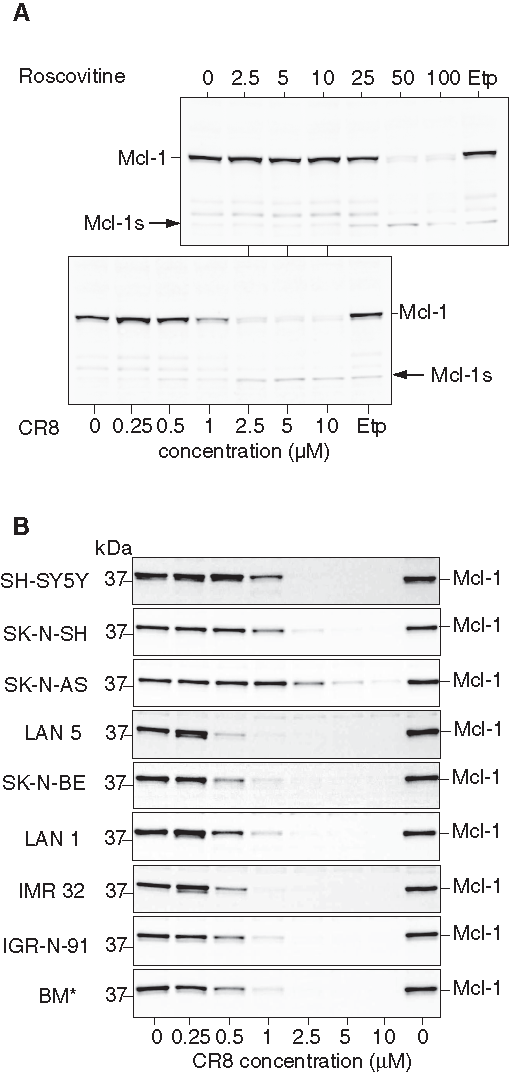
Roscovitine and CR8 induce massive down-regulation of Mcl-1 protein. (
Mcl-1 down-regulation is associated with inhibition of CDKs rather than of other targets
Besides CDKs, roscovitine and CR8 interact with a few other kinases. 22,29,44 To examine whether interaction with any of these targets might account for the induction of Mcl-1 down-regulation by roscovitine or CR8, we made use of a series of selective pharmacological inhibitors of these kinases (none of these inhibit CDKs) (Figure 4). SH-SY5Y cells were exposed to various concentrations of pharmacological inhibitors of DYRK1A (Made44 [unpublished], harmine 45 ), CK1 (IC261, 46 DRF053, 30 D4476 47 ), CLK1 (TG003 48 ), and MEK1 (UO126, PD098059 49 ). In contrast to a general kinase inhibitor (staurosporine 50 ), roscovitine, and CR8, none of these inhibitors induced Mcl-1 down-regulation. N6-methyl-roscovitine, a kinase inactive derivative of roscovitine, 51 was unable to trigger Mcl-1 down-regulation. These results suggest that CR8 and roscovitine trigger Mcl-1 down-regulation through inhibition of CDKs rather than by interaction with secondary targets.

Down-regulation of Mcl-1 is not induced by inhibitors of other kinase targets of roscovitine or CR8. SH-SY5Y cells were exposed for 10 hours to pharmacological inhibitors of DYRK1A (Made44, harmine), CK1 (IC261, DRF053, D4476), CLK1 (TG003), MEK1 (UO126, PD098059); to a general kinase inhibitor (staurosporine); to a kinase-inactive derivative of roscovitine (N6-methyl-roscovitine); or to roscovitine or CR8. All drugs were tested at 5, 15, and 50 µM, except staurosporine (0.2, 0.6, and 2 µM). Cells were harvested and proteins were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blotting with anti-Mcl-1 antibodies. α-Tubulin was used as a loading control.
CR8 directly interacts with CDK9/cyclin T at the adenosine triphosphate (ATP) binding site
Recently, we showed the key role of CDK9 inhibition in the cellular effects of roscovitine and CR8. 29 Both compounds are potent inhibitors of CDK9 (IC50 values: 0.23 µM and 0.11 µM, respectively). CDK9 inhibition results in RNA polymerase 2 inhibition, partial and transient inhibition of transcription, and down-regulation of short-lived proteins (like Mcl-1). To understand the molecular mechanism of action of CR8 and roscovitine, we previously solved their structures in complex with CDK2/cyclin A. 29 To compare the binding modes of CR8 inhibitor to CDK9/cyclin T and to CDK2/cyclin A, the structure of a CDK9/cyclin T/CR8 complex was solved (PDB: 3LQ5). CR8 is bound within the ATP binding site between the N- and C-terminal lobes of the kinase (Figure 5A). A comparison with the apo (PDB ID: 3BLH) and AMPPNP-bound (PDB ID: 3BLQ) CDK9/cyclin T structures reveals that the CDK9 C-terminal lobe is rotated with respect to the N-terminal lobe about the peptide bond linking residues Phe105 and Cys106. This movement is accompanied by the glycine loop rotating round and down to occlude the inhibitor binding site (Figure 5). Similar rearrangements to the CDK9 structure have been observed to accompany the binding of other ATP-competitive inhibitors 52 (unpublished data). (S)-CR8 adopts a similar binding mode within the CDK9 ATP binding site to that of (R)-CR8 bound to CDK2 reported previously (PDB ID: 3DDP). 29 N7 of the purine ring and the 6-amino group form hydrogen bonds to the backbone amide nitrogen and the carbonyl moiety, respectively, of Cys106 of the hinge region (Figure 5B). N7 mimics the interaction of N1 of ATP with CDK9, but the interaction made by the 6-amino group has no equivalent in the ATP-bound structure. Residues Ile25 and Leu156 sandwich the CR8 purine ring from above and below, respectively. This orientation of the inhibitor within the ATP binding site locates the phenyl-pyridine ring out of the ATP binding site where it can form favorable interactions with the side chains of Ile25, Phe105, and Ala23 that are reminiscent of the interactions between (R)-CR8 and CDK2 residues Ile10, Phe82, and Glu8 within the CDK2/cyclin A/(R)-CR8 complex structure. 29 The weaker electron density for the CR8 phenyl-pyridine moiety compared to that of the purine ring, however, suggests that there is some flexibility in the phenyl-pyridine ring location. Similarly, the electron density for the hydroxymethyl and ethyl groups is weaker, indicating additional conformational variability of these groups within the crystal.
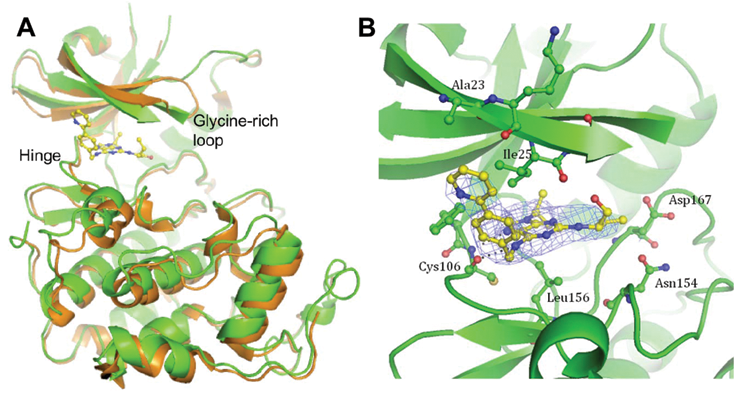
Co-crystal structure of CR8 with CDK9/cyclin T. (
Mcl-1 is a short-lived protein in neuroblastoma SH-SY5Y cells
Mcl-1 has been reported to undergo rapid turnover (reviews in references 37-39). To investigate this in the NB SH-SY5Y cell line, we made use of MG132, a proteasome inhibitor, 53 or cycloheximide, a general inhibitor of protein synthesis (Figure 6). Exposure to MG132 led to a rapid and massive accumulation of Mcl-1 protein, which was only partially moderated by co-treatment with roscovitine or CR8 (Figure 6A). In contrast, cycloheximide treatment led to a rapid down-regulation of Mcl-1 protein level, which was not accelerated by co-treatment with roscovitine or CR8 (Figure 6B). PARP cleavage measured in the same experiment followed the same pattern as Mcl-1 down-regulation (Suppl. Fig. S2) (although MG-132 alone induced late PARP cleavage). These results suggest that roscovitine or CR8 trigger Mcl-1 down-regulation primarily by inhibiting its synthesis than by increasing its degradation. Interestingly, in the presence of MG132 (Figure 6) and in some neuroblastoma cell lines more than others (Figure 3B), Mcl-1 appears as a doublet. CR8 and roscovitine trigger a motility shift (faster migration), suggesting that Mcl-1 is phosphorylated by a kinase that is inhibited by the two drugs.
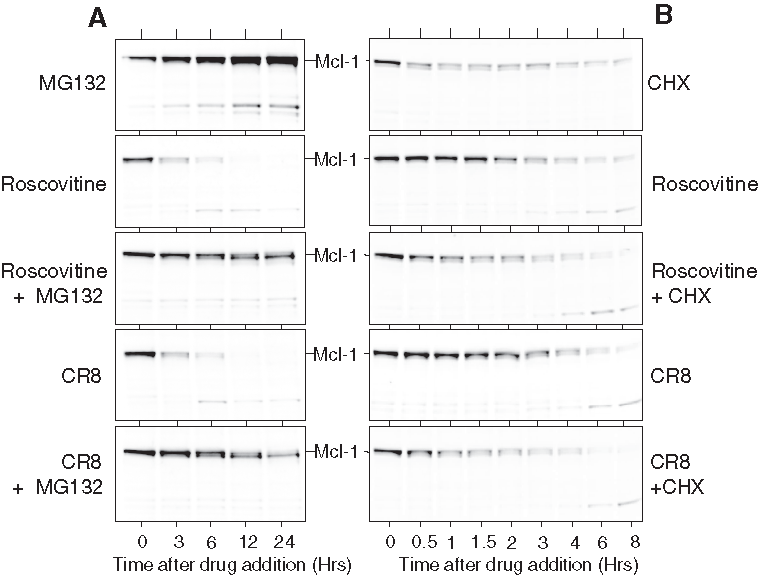
Proteasome inhibition (
Mcl-1 down-regulation induced by roscovitine and CR8 is independent of p53 up-regulation or MYCN amplification
Induction of p53 by roscovitine has been reported in numerous cell lines. 54-60 A similar p53 up-regulation was observed in SH-SY5Y cells exposed to roscovitine and CR8 (Figure 7A). Time- and concentration-dependent effects and higher potency of CR8 over roscovitine were observed. At 25 µM roscovitine and 1 µM CR8, p53 was maximally up-regulated. However, at these respective concentrations, no or little Mcl-1 down-regulation was observed. To further investigate the potential link between p53 up-regulation and Mcl-1 down-regulation, we made use of nutlin-3, an inhibitor of the p53/Hdm2 interaction 61 (Figure 7B). Exposure to this compound leads to stabilization and accumulation of p53 and Hdm2. 62 However, despite p53 up-regulation, Mcl-1 levels remained constant (Figure 7B). The use of various NB cell lines showed that Mcl-1 down-regulation was induced by roscovitine and CR8 (Figure 3B), even in cells where p53 was absent (Lan1), mutated (SK-N-BE), or had undergone a major increase in size as a result of the duplication of 3 exons (IGR-N-91, BM*) (Table 1). Finally, we used p53−/−, p53+/+, and wild-type HCT116 cells. 63 Both roscovitine and CR8 were able to trigger Mcl-1 down-regulation, even in the complete absence of p53 (Suppl. Fig. S3). Taken together, these results suggest that p53 or p53 up-regulation is dispensable for Mcl-1 down-regulation induced by roscovitine and CR8.
Mcl-1 down-regulation induced by roscovitine and CR8 is independent from p53 induction. (
MYCN amplification is observed in HR-NB. Mcl-1 down-regulation was observed in all NB cell lines tested, whether MYCN was amplified (LAN 1, LAN 5, SK-N-BE, IMR 32, IGR-N-91, BM*) or not (SH-SY5Y, SK-N-SH, SK-N-AS) (Figure 3B; Table 1). The less sensitive cell lines (in terms of Mcl-1 down-regulation), SK-N-AS and SK-N-SH, displayed no MYCN amplification. These results suggest that neither Mcl-1 expression nor its down-regulation triggered by the CDK inhibitors depend on MYCN overexpression.
Roscovitine and CR8 induce a transient increase in free Noxa level
The main function of Mcl-1 is to titer apoptosis-inducing factors such as Noxa and Puma, thereby favoring cell survival over cell death. 64 To analyze the role of Noxa in SH-SY5Y cells, we exposed cells for various durations to the CDK inhibitors. Total Mcl-1 and total Noxa were detected by Western blotting. Both were down-regulated as a result of exposure to CR8 (Figure 8A,B). Mcl-1 was immunoprecipitated with specific antibodies, and its associated Noxa was estimated by Western blotting. Mcl-1-bound Noxa was down-regulated by CR8 (Figure 8C). The supernatants of the Mcl-1 immunoprecipitates were then exposed to Noxa-specific antibodies, allowing the recovery of free Noxa (i.e., not associated with Mcl-1). This free Noxa fraction showed a transient increase in its level (Figure 8D). These results suggest that while Mcl-1 is down-regulated, the free Noxa level increases momentarily. This free Noxa fraction, as well as the proapoptotic Mcl-1 fragments, may be the actual triggers for apoptotic death in NB cells.
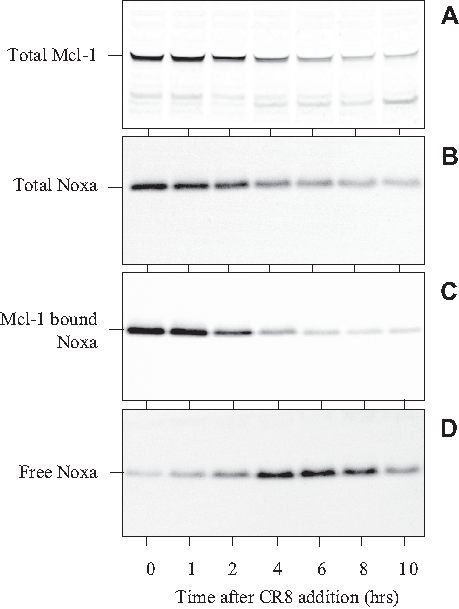
CR8 triggers transient increase in free Noxa level. SH-SY5Y cells were exposed to 5 µM CR8 at time 0, and cell samples were collected at various times thereafter. Proteins were extracted and resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blotting with antibodies directed against Mcl-1 (
Discussion
Neuroblastoma is a complex disease for which a substantial fraction of its young patients show very poor prognosis (HR-NB). Although the molecular signature of HR-NB is being investigated in detail, 5,65,66 as a result of its complexity, this analysis has only provided markers to categorize patients rather than validated targets to treat them. Nevertheless, a few such potential targets are starting to emerge, such as MYCN, 67-70 ALK (anaplastic lymphoma kinase), 71,72 and MDM2 73 (review in Wagner and Danks 7 ). A recent article 41 suggests that Mcl-1 may be playing a crucial role in NB resistance to chemotherapy and tumor cell survival. First, a correlation was observed between high expression of Mcl-1 and Bcl-2 and high-risk phenotype. 41,74 We have confirmed the MYCN status-independent expression of Mcl-1 in clinical samples of NB stages 1, 2, 3, 4, and 4S by Western blotting (data not shown). Second, Mcl-1 RNA interference induced down-regulation of Mcl-1 protein levels in HR-NB cell lines, leading to induction of apoptosis. 41 Third, NB cells’ resistance to cytotoxic chemotherapy or Bcl-2 antagonists was reduced by Mcl-1 silencing. 41 Finally, the Mcl-1 gene maps to 1q21, a region that is frequently seen in high copy number in many NB cell lines and primary NB tumors, and is associated with poor prognosis. 6,75 Altogether, these data suggest that down-regulating Mcl-1 expression would be favorable for the treatment of HR-NB. This is probably true for an increasing number of tumor types. 40
In this article, we report that roscovitine and CR8, two purine CDK inhibitors, trigger rapid and massive down-regulation of the critical survival factor Mcl-1 in all 9 NB cell lines tested. This effect is rapid and marked, and it occurs independently of MYCN and p53 status. In other words, HR-NB cell lines are thus as sensitive to the CDK inhibitors as NB cell lines in which MYCN is not amplified and p53 expression is normal.
Our current hypothesis (Figure 9) is that roscovitine and CR8 inhibit CDK9/cyclin T and CDK7/cyclin H, two kinases the activity of which is indispensable for RNA polymerase 2 activity. Transient inhibition of RNA polymerase 2 results in transient inhibition of transcription, which primarily impinges on high-turnover mRNAs and short-lived proteins. Mcl-1 is clearly such a case, and indeed, Mcl-1 mRNA and Mcl-1 protein levels are rapidly down-regulated. Those cells that rely on Mcl-1 for their balance between survival and cell death are thus tilted toward cell death. Such is the case for NB cell lines. Support for this hypothetical mechanism of action comes from a series of results reported in this work and in a previous article 29 :
CDK9 and CDK7 are among the most sensitive targets of roscovitine and CR8. 29 Co-crystallization of CDK9/cyclin T with CR8 confirms the excellent fit between the inhibitor and the ATP binding pocket of CDK9 (Figure 5). However, the contribution of other kinase and nonkinase targets to Mcl-1 down-regulation and NB cell death cannot be formally ruled out, despite the fact that well-characterized inhibitors of some of these targets (CK1, DYRKs, CLKs, Erks) have no effects on NB MCl-1 down-regulation (Figure 4). In particular, the role of CDK1, CDK2, and CDK5 inhibition remains to be clarified.
Inhibition, by roscovitine and CR8, of RNA polymerase 2 phosphorylation on serine 9, its CDK9 site, has been demonstrated. 29
A global transcriptomics and proteomics study has shown that roscovitine and CR8 trigger down-regulation of a selection of transcripts, including that of Mcl-1 and of other selected proteins (Delehouzé et al., in preparation). Detailed analysis reported herein shows that both CDK inhibitors down-regulate, in a concentration- and time-dependent manner, the expression of Mcl-1 mRNA (Figure 2) and protein (Figure 3).
Mcl-1 down-regulation allows a transient increase in free Noxa concentration (Figure 8). We suspect this factor plays the major role in cell death induction, which is likely to occur in a Bax/Bak-dependent manner, as demonstrated recently for roscovitine 76 and CR8 (Garrofé-Ochoa et al., data not shown) using Bax−/−/Bak−/− fibroblasts.
The kinetics of PARP cleavage (Suppl. Fig. S2) closely parallel those of Mcl-1 down-regulation and free Noxa accumulation, further supporting the model.
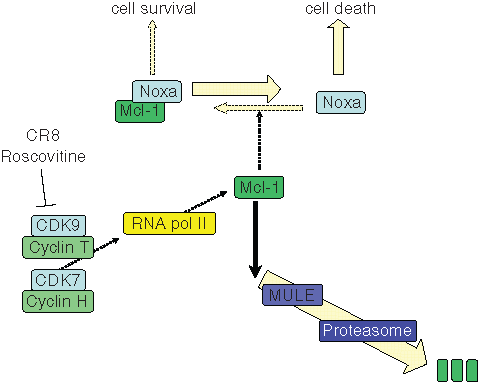
Hypothetic scheme of cell death induction pathway triggered roscovitine and CR8 in human neuroblastoma (NB) cells. In growing NB cells, the cell survival/cell death balance is maintained by the Bcl-2 family member Mcl-1, a survival factor that binds to and neutralizes proapoptotic proteins such as Noxa. Mcl-1 is a short-lived protein being constantly synthesized (through RNA polymerase 2, itself under the control of CDK7/cyclin H and CDK9/cyclin T) and constantly degraded (through the Mcl-1 ubiquitin ligase [MULE] and the proteasome). The CDK inhibitors roscovitine and CR8 interact with CDK7 and CDK9, preventing phosphorylation of RNA polymerase 2 at Ser-2 and Ser-9, respectively. Consequently, mRNA synthesis is inhibited. Short-lived mRNAs and short-lived proteins are down-regulated. This is the case for Mcl-1. Reduction of Mcl-1 protein level results in a transient increase in free Noxa protein, which triggers apoptotic cell death through a Bax/Bak-dependent pathway. Although p53 may be up-regulated, it is not involved in Mcl-1 down-regulation and cell death.
The difference between roscovitine and CR8 in their inhibitory activity on kinase targets is rather small (e.g., their IC50 values for CDK9 are 0.23 and 0.11 µM, respectively). Why CR8 is 100 to 200 times more potent than roscovitine at inducing Mcl-1 down-regulation and cell death remains unexplained. There might be subtle differences in cell permeability, intracellular stability, turnover, distribution, or binding to irrelevant “sink” structures. In addition, an unidentified target not shared by roscovitine and CR8 might either enhance the effects of CR8 or titer down the effects of roscovitine. Alternatively, the improved efficacy of CR8 over roscovitine may rely on the additive effects of minor improvements on a wide set of targets rather than a single unidentified target.
Altogether, these results support the notion that CDK inhibitors, by down-regulating the survival factor Mcl-1, could constitute a promising approach for the treatment of neuroblastoma, especially MYCN-amplified, high-risk neuroblastoma.
Materials and Methods
Reagents; Antibodies
Roscovitine, N6-methyl-roscovitine, CR8, and DRF053 were synthesized as previously described. 30,43,51 Harmine, IC261, D4476, UO126, and PD098059 were purchased from Sigma (St Quentin Fallavier, France). Made44 and TG003 were gifts from, respectively, Dr. J. P. Bazureau and F. Carreaux (University of Rennes, France) and from Dr. M. Hagiwara (Tokyo Medical and Dental University, Japan). Compounds were stored dry but diluted as 10 mM stock solutions in dimethylsulfoxide (DMSO) prior to use.
Cell Titer 96® containing the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) reagent was purchased from Promega (Madison, WI, USA). The protease inhibitor cocktail was from Roche (Penzberg, Germany). Unless otherwise stated, the nonlisted reagents were from Sigma.
Monoclonal antibodies against actin and tubulin were from Calbiochem (Madison, WI, USA) and Santa Cruz Biotechnology (Santa Cruz, CA, USA), respectively. Polyclonal antibodies against Mcl-1 and Noxa or monoclonal antibody against p53 and Hdm2 were obtained from Santa Cruz Biotechnology.
Homogenization buffer
60 mM β-glycerophosphate, 15 mM p-nitrophenyl phosphate, 25 mM MOPS (pH 7.2), 15 mM EGTA, 15 mM MgCl2, 1 mM dithiothreitol (DTT), 1 mM sodium vanadate, 1 mM NaF, 1 mM phenylphosphate, 0.1% Nonidet P-40, and protease inhibitor cocktail.
Expression, purification, and co-crystallization of human CDK9/cyclin T with CR8
X-ray crystallography data collection and processing, structure solution, and refinement. CDK9 (residues 1-330)/cyclin T (residues 1-259) were expressed and purified as described previously. 52 Protein was concentrated to 4 mg/mL in buffer containing 20 mM Tris (pH 8.0), 500 mM NaCl, 10% glycerol, and 5 mM DTT, and crystals were grown at 4°C in sitting drops using 14% PEG 1000, 100 mM NaK-phosphate (pH 6.2), 500 mM NaCl, and 4 mM TCEP as the precipitant solution. Crystals were soaked in the same solution containing 1 mM inhibitor for 4 hours before buffer exchange into cryo-protectant (the precipitant solution supplemented with 15% glycerol and 1 mM CR8) and cryo-cooled in liquid nitrogen. Data were collected at the Diamond Light Source, Beamline I02 from a single crystal at 100K and processed using MOSFLM and SCALA. 77 Strong electron density was observed corresponding to the inhibitor after initial rigid body refinement using phenix.refine 78 and a refined CDK9/cyclin T structure (PDB code 3BLH) as a template. The initial model was rebuilt in COOT 79 and refined using phenix.refine. The final model was validated using molprobity. 80 Statistics of the data set used and of the refined structures are provided in the Supplementary Material section (Table S1). Coordinates of the co-crystal structures are available at the PDB code 3LQ5.
Cell lines and culture conditions; cell survival quantification
All 9 NB cell lines (molecular characteristics in Table 1) were grown in Dulbecco’s modified Eagle’s medium (DMEM) from Invitrogen (Cergy Pontoise, France), with the exception of LAN 1, LAN 5, and IMR 32, which were grown in RPMI (Invitrogen). The HCT116 human colorectal carcinoma cell lines (kindly provided by Dr. B. Vogelstein, Baltimore, MD, USA) were grown in McCoy’s 5a medium from ATCC (Manassas, VA, USA). All media were supplemented with antibiotics (penicillin-streptomycin) from Lonza (Levallois Perret, France) and 10% volume of fetal calf serum from Invitrogen. Cells were cultured at 37°C with 5% CO2. Drug treatments were performed on exponentially growing cultures at the indicated time and concentrations. Control experiments were carried out using appropriate dilutions of DMSO (maximum 1% DMSO). Cell viability was determined by measuring the reduction of MTS as described in Ribas and Boix. 81
Immunoprecipitation
Antibodies (Mcl-1 or Noxa) were prebound to protein A-Sepharose beads (Pierce, Rockford, IL, USA) for 1 hour in 1 mL of homogenization buffer supplemented with 1% bovine serum albumin (BSA). Immunoprecipitations were performed with 1 or 1.5 mg proteins during 1 hour at 4°C. Beads were washed 4 times with bead buffer (50 mM Tris [pH 7.4], 5 mM NaF, 250 mM NaCl, 5 mM EDTA, 5 mM EGTA, 0.1% Nonidet P-40, and protease inhibitor cocktail) before addition of 2X sample loading buffer. Following heat denaturation for 3 minutes, the bound proteins were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting.
Electrophoresis and Western blotting
Cells were resuspended, lysed for 30 minutes at 4°C in homogenization buffer, and sonicated. After centrifugation (17,000 g for 15 minutes at 4°C), the protein concentration was determined in the supernatants by the DC protein assay (Bio-Rad, Hercules, CA, USA). Following heat denaturation for 3 minutes, proteins were separated by 10% or 7%, according to protein size, NuPAGE precast Bis-Tris or Tris-Acetate polyacrylamide mini gel electrophoresis (Invitrogen) with MOPS SDS running buffer. Proteins were transferred to 0.45-µm nitrocellulose filters (Schleicher & Schuell, Whatman, Dessel, Germany). These were blocked with 5% low-fat milk in Tris-buffered saline/Tween-20, incubated for 1 hour with antibodies (anti-actin: 1:2000) or overnight at 4°C (Mcl-1 [1:500], Noxa [1:500], p53 [1:1000], Hdm2 [1:500], tubulin [1:500]), and analyzed by Enhanced Chemiluminescence (ECL; Amersham, Les Ulis, France).
Polymerase chain reaction amplification of Mcl-1 RNA
Total RNA from SH-SY5Y cells was extracted using RNeasy Plus Mini kit (Qiagen S.A., Courtaboeuf, France) according to the manufacturer’s instructions. Contaminating genome DNA was eliminated during the extraction. Then, 1 µg of total RNA was reverse transcribed using the Omniscript Reverse Transcription kit (Qiagen) according to the manufacturer’s instructions. Expression level of Mcl-1 mRNA was detected by PCR using the HotStarTaq PCR kit (Qiagen). Expression level of actin mRNA was evaluated as an internal control as an indication of equal amounts of RNA in each reaction. The oligonucleotide sequences were as follows. Actin forward: 5′-GGA-CTT-CGA-GCA-AGA-GAT-GG-3′; actin reverse: 5′-AGC-ACT-GTG-TTG-GCG-TAC-AG-3′; Mcl-1 forward: 5′-TAA-GGA-CAA-AAC-GGG-ACT-GG-3′; Mcl-1 reverse: 5′-ACC-AGC-TCC-TAC-TCC-AGC-AA-3′. The PCR conditions were 95°C for 15 minutes, 28 cycles of amplification (94°C for 30 seconds, 42°C for 30 seconds, 72°C for 1 minute), and 72°C for 10 minutes.
Footnotes
Acknowledgements
The authors are grateful to Dr. B. Vogelstein for the HCT116 cell lines and Dr. J.P. Bazureau, Dr. F. Carreaux, and Dr. M. Hagiwara for reagents.
K. Bettayeb, N. Oumata, H. Galons, and L. Meijer are coinventors on a patent disclosing CR8. L. Meijer is an inventor on a patent disclosing roscovitine.
This research was supported by grants from “Association pour la Recherche sur le Cancer” (ARC-1092) (LM), the EEC (FP6-2002-Life Sciences & Health, PRO-KINASE Research Project) (LM), the “Ligue Nationale contre le Cancer” (LM), the “Cancéropole Grand-Ouest” grant (LM), the “Institut National du Cancer” (INCa « Cancer Détection d’innovations 2006 ») (LM), the “Association France-Alzheimer Finistère” (LM), “Enfants et Santé/SFCE (JBd),” and “Comité Montbéliard Ligue contre le Cancer” (JBd). KB was supported by a fellowship from the “Ministère de la Recherche” and from the “Association pour la Recherche sur le Cancer.” DB was supported by the FP6–Marie Curie Action–ESTeam MEST-CT-2005-020737 program. NO benefited from a PhD fellowship from the “Institut National du Cancer.” The authors are grateful to the beamline scientists at the Diamond Light Source for excellent facilities and M. Noble for his helpful comments. This research was supported by grants from the Wellcome Trust (AH) and the Medical Research Council (SB and JAE). Coordinates of the CDK9/cyclin T/CR8 crystal structure are available under PDB file 3LQ5.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.