
Abstract
Innate resistance to various therapeutic interventions is a hallmark of cancer. In recent years, acquired resistance has emerged as a daunting challenge to targeted cancer therapy, which abolishes the efficacy of otherwise successful targeting drugs. Cancer cells gain the resistance property through a variety of mechanisms in primary and metastatic cancers, involving cellular intrinsic and extrinsic factors. Increasing evidence suggests that the mammalian stress response gene sirtuin 1 (SIRT1) plays a critical role in multiple aspects of cancer drug resistance. SIRT1 decreases drug penetration, confers proliferation and antiapoptotic survival advantages to cancer cells, facilitates acquired resistance through genetic mutations, promotes the survival of cancer stem cells, and changes the tumor microenvironment for resistance in cell-autonomous and -nonautonomous manners. This article provides an overview of research advances in the roles of SIRT1 in cancer drug resistance and highlights the prospect of targeting SIRT1 as a new strategy to overcome cancer drug resistance and improve therapeutic outcomes.
Keywords
Introduction
With decades of mechanistic studies of cancer development, cancer treatment has advanced from using general cytotoxic agents to molecular target–based smart therapy. 1 However, cancer drug resistance remains a major challenge for successful treatment. Cancer drug resistance comprises primary (innate) and secondary (acquired) resistance in response to treatment and is regulated by multiple mechanisms involving intrinsic and extrinsic factors. Innate resistance refers to cancer’s inherent ability to be refractory to drugs. Traditionally, innate resistance is known to be mediated by several mechanisms including 1) reduced drug intake through mutations or loss of drug transporters, enhanced drug efflux by overexpressing ATP-binding cassette (ABC) transporters such as ABCB1 (also known as multidrug resistance protein 1 [MDR1]) and multidrug resistance-associated proteins, and incomplete drug penetration inside solid tumors2-4; 2) activated drug metabolism and detoxification system such as cytochrome P450 (CYP450), superoxide dismutase (SOD), and glutathione S-transferase (GST)5,6; 3) gain of function of antiapoptosis and cell cycle checkpoint-evading mechanisms 7 ; and 4) activation of DNA repair machineries to reduce drug-induced DNA damage. 2
Acquired resistance refers to cancer resistance developed after initial remission as a result of the treatment, with the relapsed disease no longer responding to the initial drug. Acquired resistance may thus be considered an adaptive or evolutionary outcome of cancer cells to the lethal action of the therapeutic agents. Similar to innate resistance, acquired resistance can be mediated by multiple mechanisms including genetic mutations and gene amplification and some of which are also involved in innate resistance. 8 One prominent mechanism that plagues targeted cancer therapy is the acquisition of resistant genetic mutations, abolishing the efficacy of the targeting drugs. 1 Molecular mechanisms of acquired resistance through mutation acquisition are poorly understood, but recent studies have started to shed novel insight on this aspect and will be discussed in this review. Furthermore, cancer stem cells and the tumor microenvironment have increasingly been recognized as important factors driving innate and/or acquired resistance.
Sirtuin 1 (SIRT1) is a mammalian nicotinamide adenine dinucleotide (NAD+)–dependent lysine deacetylase.9,10 SIRT1 belongs to class III histone deacetylases (HDACs) that are structurally and functionally distinct from class I, II, and IV HDACs. There are 7 sirtuin family members, and among them, SIRT1 has the highest homology to the founding member of NAD-dependent lysine deacetylases, yeast silent information regulator 2 (Sir2), that is associated with life-span extension upon calorie restriction. 11 Extensive studies over the past decade have established that SIRT1 is involved in the regulation of a wide variety of biological functions including gene expression, cell survival, proliferation, differentiation, metabolism, immune response, and carcinogenesis. These broad regulatory functions are likely a result of central roles of SIRT1 in metabolic, oxidative, genotoxic, and oncogenic stress responses as reviewed previously.12-14 SIRT1 is overexpressed in many types of solid tumors and hematopoietic malignancies.15-20 Although it has been an issue of debate whether SIRT1 is an oncogene or a tumor suppressor, increasing evidence suggests that SIRT1 is a major player in cancer drug resistance. This review will focus on the roles of SIRT1 in cancer drug resistance and highlight the potential prospects of targeting SIRT1 to overcome resistance and improve cancer treatment.
Overview of Regulation Networks of SIRT1
SIRT1 is subjected to multiple layers of regulation from transcription and translation to protein functions in cancer cells (Fig. 1). First, the transcriptional factors hypermethylated in cancer 1 (HIC1) and p53 repress SIRT1 transcription,21,22 whereas E2F1, c-Myc, N-Myc, and signal transducer and activator of transcription 5 (STAT5) activate SIRT1 expression.20,23-26 Feedback regulation loops are typically found with these transcriptional regulators, suggesting that SIRT1 transcription is under tight control to prevent constitutive activation. The recently identified SIRT1 activation by STAT5 bridges SIRT1 to cytokine and growth factor signaling 26 that may be relevant to certain physiological settings. Second, at the translational level, the RNA binding protein HuR stabilizes SIRT1 mRNA by binding to the 3′ untranslated region of SIRT1 mRNA, 27 whereas microRNA miR-34a, miR-199a, and miR-200a target SIRT1 mRNA to inhibit its translation.28-30 Third, at the posttranslational level, SIRT1 protein stability and activity are modulated by covalent modifications including phosphorylation, sumoylation, and ubiquitination. SIRT1 phosphorylation at threonine 530 and serine 540 by cell cycle–dependent kinase cyclin B/CDK1 controls cell proliferation and cell cycle profiles. 31 The dual-specificity tyrosine phosphorylation–regulated kinases DYRK1A and DYRK3 phosphorylate threonine 522 and increase SIRT1 activity to promote cell survival. 32 SIRT1 phosphorylation at serine 27 by c-Jun N-terminal kinase 2 (JNK2) stabilizes the protein, 33 whereas SIRT1 phosphorylation at serine 47 by JNK1 facilitates ubiquitination-mediated degradation. 34 Under genotoxic stress, nuclear desumoylase SENP1 removes SIRT1 sumoylation and reduces its deacetylase activity. 35 In addition, SIRT1 activity is regulated by noncovalent modifications by cellular factors and small molecules. Deleted in breast cancer 1 (DBC1) suppresses SIRT1 activity by binding to the SIRT1 catalytic core, 36 in competition with the SIRT1 C-terminal intramolecular “on-switch” peptide that activates the deacetylase function. 37 Active regulator of SIRT1 (AROS) positively regulates SIRT1 activity through direct interaction with the N-terminus of the SIRT1 protein. 38 Finally, intracellular levels of NAD+ and nicotinamide (NAM) directly affect the activity of SIRT1 since its deacetylase activity is NAD+ dependent, and NAM is the endogenous inhibitor of SIRT1. 39 It has been shown that mammalian NAD+ salvage biosynthesis enzyme nicotinamide phosphoribosyltransferase (NAMPT) is concomitantly activated with SIRT1 in several types of cancer cells to reduce the accumulation of cellular NAM and to provide sufficient NAD+ for SIRT1 functions.24,40 Numerous synthetic compounds have been made to either increase or inhibit SIRT1 activity. 41
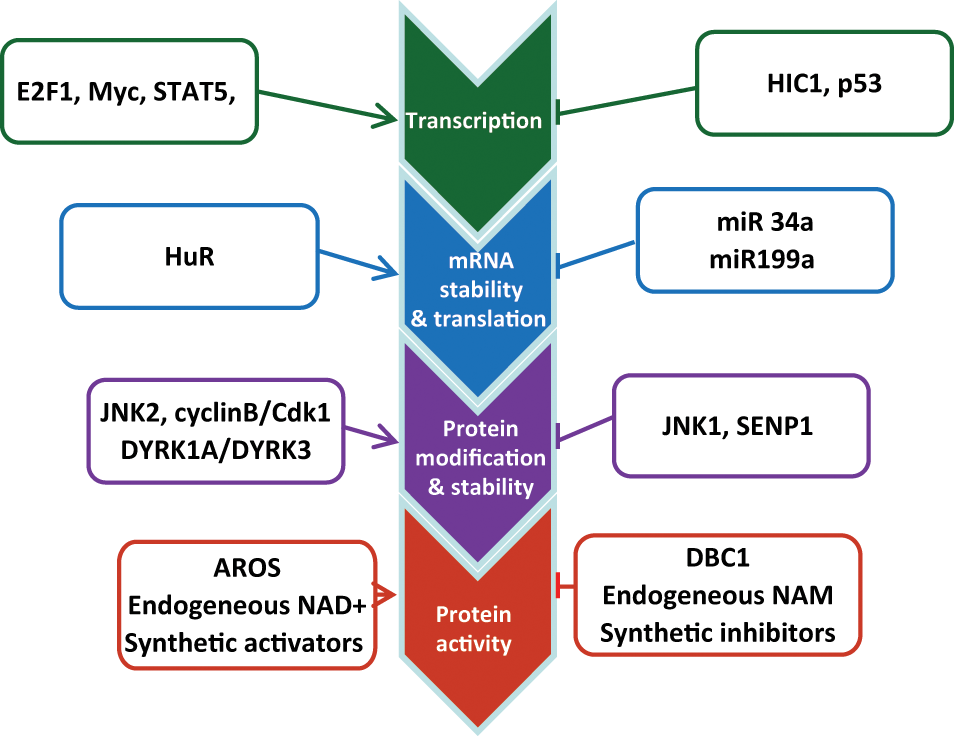
Regulation of SIRT1 expression and enzymatic activity. SIRT1 is subjected to multiple layers of regulation from transcription and translation to protein functions in cancer cells as detailed in the text.
The downstream effectors of SIRT1 include both histone and nonhistone substrates. SIRT1 directly reduces acetylation levels of histone H1 K26, H3 K9 and K14, and H4 K16.10,42,43 SIRT1 also indirectly regulates histone methylation through interacting with other epigenetic enzymes such as methyltransferase SUV39H1. 44 The alteration of histone codes may effectively change the chromatin structure and gene transcription involved in carcinogenesis and cancer drug resistance. The nonhistone substrates of SIRT1 account for many biological functions that SIRT1 regulates, and they include numerous molecules in various species: 1) the transcriptional factors p53, FOXO1, FOXO3a, NF-κB, c-Myc, N-Myc, E2F, PTEN, HIF-1α/HIF-2α, PPARγ, and PGC1α; 2) histone-modifying enzymes SUV39H1, p300, TIP60, and PCAF; 3) DNA repair machinery elements Ku70, NBS1, APE1, XPA/C, and WRN; 4) nuclear receptor genes ERα, AR, and LXR; and 5) signaling molecules β-catenin and SMAD7, as detailed in previous reviews.12,14
SIRT1 Changes Drug Penetration Properties of Cancer Cells
SIRT1 regulates multiple aspects of cancer drug resistance (Fig. 2). Drug penetration is the first step of cancer therapy and is determined by absorption, distribution, metabolism, and excretion (ADME) properties in the body level and cellular level. Cellular drug intake, efflux, and detoxification reflect the ADME properties of cancer cells. Elevated expression of ABC family membrane transporters is a well-known mechanism of multidrug resistance.2,4 Chu et al. 45 and Oh et al. 46 reported that SIRT1 is activated in multiple drug-resistant cancer cell lines and clinical biopsies. Activation of SIRT1 increases the expression of ABC transporter P-glycoprotein and MDR1 through deacetylating FOXO1 and increasing the nuclear level of FOXO1. Li et al. 47 showed that SIRT1 positively regulates nuclear receptor liver X receptor (LXR) and increases the expression of the LXR target gene ABCA1, another ABC transporter. Consequently, the activated expression of ABC transporters by SIRT1 can increase drug efflux, decrease drug concentration in cancer cells, and lead to drug resistance.
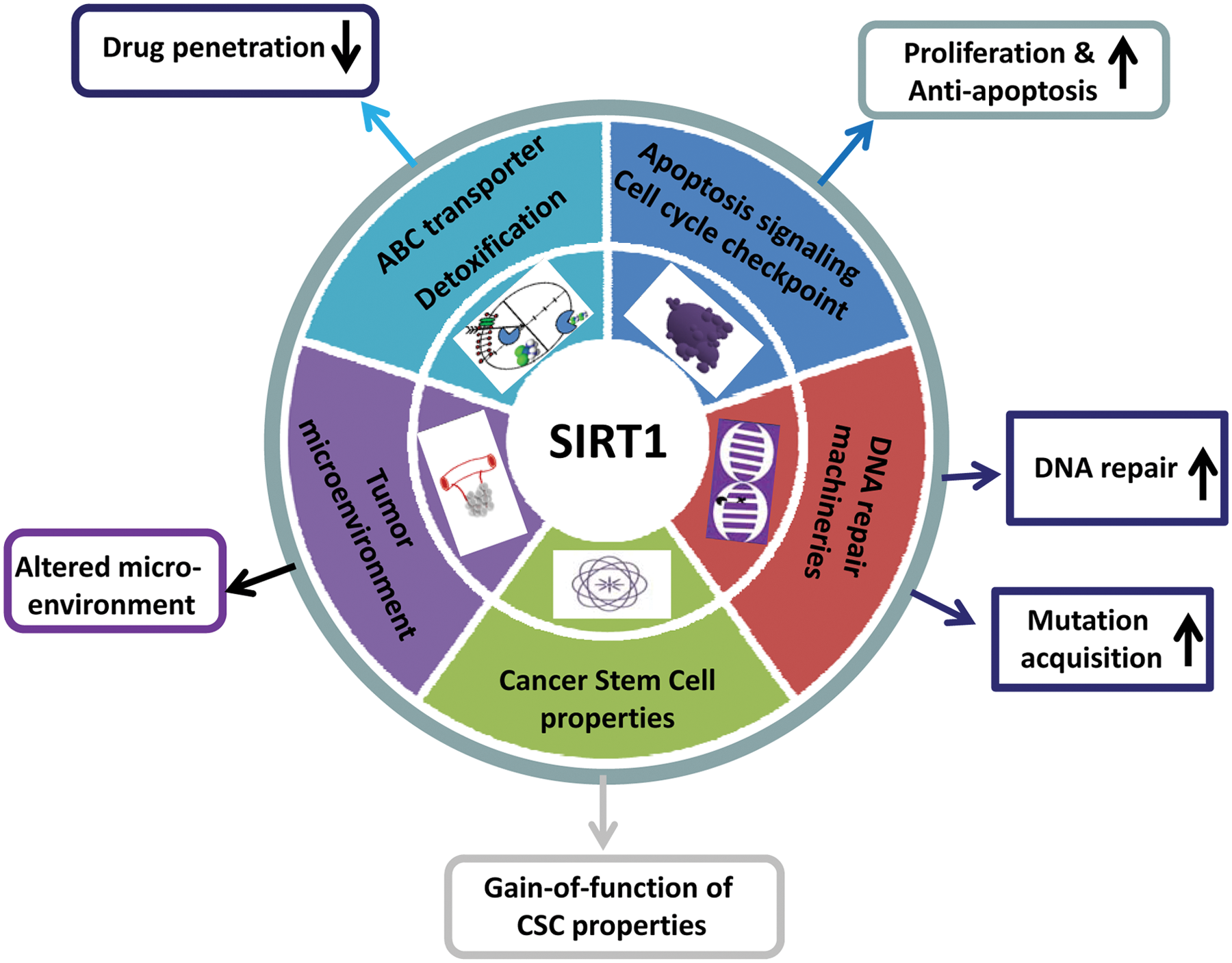
SIRT1 regulates multiple pathways of cancer drug resistance. Overexpression of SIRT1 in cancer cells reduces drug penetration, confers cell proliferation and antiapoptotic advantages, promotes DNA damage repair and acquisition of genetic mutations under therapeutic stress, increases the gain of function of cancer stem cell properties, and modulates the tumor microenvironment for cancer cell drug resistance.
Under stress conditions, SIRT1 activates cellular detoxification systems. For example, SIRT1 activates FOXO3a and increases the expression of manganese SOD (MnSOD) mRNA and protein, which contributes to cellular resistance to oxidative stress.48,49 Similarly, activation of SIRT1 in breast cancer cells activates SOD and glutathione peroxidase (Gpx) against oxidative stress. 50 However, there is no report yet whether SIRT1 can activate CYP450 and GST, the major cellular drug detoxification systems, to decrease drug accumulation and cellular damage in cancer cells.
SIRT1 Confers Proliferative and Antiapoptotic Advantages to Cancer Cells
SIRT1 expression is elevated in a large spectrum of cancers. SIRT1 deacetylates several master transcriptional factors that are involved in the regulation of cell apoptosis and senescence. Activation of SIRT1 deacetylates p53/p73 and inhibits p53/p73-induced apoptosis,51-53 tips FOXO-dependent responses away from apoptosis and towards stress resistance,48,49 and inhibits cell cycle and apoptosis regulator E2F1 activities to decrease the cellular sensitivity to DNA damage. 23 In chronic myeloid leukemia (CML), SIRT1 is transcriptionally activated by BCR-ABL in hematopoietic stem/progenitor cells in part through STAT5 and promotes BCR-ABL transformation and leukemogenesis.26,54 SIRT1 promotes CML cell survival and proliferation in association with deacetylation of its substrates p53 and Ku70. SIRT1 also activates antiapoptosis factors such as BCL6 for the survival of lymphoma cells. 55
SIRT1 Enhances DNA Damage Repair in Cancer Cells
Inducing apoptosis by increasing DNA damage has been explored as a means to selectively kill cancer cells that harbor certain defects in DNA damage repair.56,57 However, by boosting alternative DNA repair and reducing damage, cancer cells could bypass apoptosis and develop resistance. Upon DNA damage, SIRT1 relocates to DNA breaks to promote repair and cell survival.58,59 SIRT1 enhances the functions of multiple repair pathways including nonhomologous end joining (NHEJ) repair, homologous recombination (HR) repair, base excision repair (BER), and nucleotide excision repair (NER). SIRT1 regulates these pathways through deacetylating Ku70,59-61 Nijmegen breakage syndrome protein (NBS1), 25 apurinic/apyrimidinic endonuclease-1 (APE1), 62 xeroderma pigmentosum group A/C (XPA/XPC),63,64 and Werner syndrome protein. 65 As a result, inhibition of SIRT1 can sensitize cancer cells to several types of DNA damage–inducing agents.46,66,67
SIRT1 Promotes Acquired Resistance through Genetic Mutations
Acquisition of genetic mutations is a major mechanism underlying cancer acquired resistance with different molecular targets in a variety of cancers including CML,68,69 lung cancer,70-74 colon cancer,75,76 ovarian and breast cancer,77,78 and gastrointestinal cancer. 79 The conventional explanation is that mutations form spontaneously and randomly before cancer has undergone chemotherapy, and these rare pre-existing mutations may be selected for resistance under chemotherapy. However, precise molecular mechanisms of how resistant mutations are actually acquired during cancer therapy are largely unknown.
Dissecting mechanisms of cancer acquired resistance would be difficult without a good modeling system in mammalian cells. 80 Recently, Yuan et al. 81 have developed a novel culture model of CML acquired resistance based on a blast crisis CML cell line KCL-22. CML is a hematopoietic stem cell malignancy caused by oncogenic fusion gene BCR-ABL that confers multiple advantages of cell proliferation and alters DNA damage repair machineries. 82 Although the development of BCR-ABL tyrosine kinase inhibitor (TKI) imatinib mesylate significantly advances CML treatment, 83 acquired resistance primarily mediated by genetic mutations of BCR-ABL remains a challenge, in particular, for the advanced phases of the disease.68,69,84 Mutations of BCR-ABL change the conformation of the protein and impair the binding of TKIs to the BCR-ABL kinase domain, which leads to drug resistance and CML relapse.85,86 In the KCL-22 cell model, following initial apoptotic response, T315I BCR-ABL mutation is rapidly acquired upon treatment with therapeutic concentrations of imatinib, 81 recapitulating features of clinical BCR-ABL mutation acquisition. Interestingly, most, if not all, BCR-ABL mutations are acquired de novo in this model, suggesting a previously underestimated route of mutation acquisition that may contribute to the faster relapse of cancer patients under therapeutic stress. 87
By using the KCL-22 cell model, Wang et al. 88 showed that SIRT1 is a critical factor for promoting the acquisition of genetic mutations of BCR-ABL for CML resistance. SIRT1 gene knockdown or inhibition by small molecule inhibitors blocks the acquisition of BCR-ABL mutations in CML cells and prevents CML cell relapse from TKIs. SIRT1 affects mutation acquisition not only on the BCR-ABL gene but also on other genes. SIRT1 inhibition efficiently inhibits de novo mutation acquisition of the hypoxanthine phosphoribosyltransferase (HPRT) gene in CML and prostate cancer cells upon treatment with the chemotherapeutic agent camptothecin. In contrast to DNA damage induction by camptothecin, imatinib does not act through enhancing DNA damage, 81 and suppression of mutation acquisition by SIRT1 inhibition does not require increased cell killing. 88 Wang et al. 88 showed that SIRT1 promotes mutation acquisition in association with its ability to enhance error-prone NHEJ DNA damage repair through deacetylating Ku70. Inhibition of Ku70 or HR repair factors NBS and RAD51 all suppresses BCR-ABL mutations, with the latter apparently impacting NHEJ activity.
The study by Wang et al. 88 contrasts with our conventional thinking that increased DNA damage repair should lead to reduced mutations. This surprise may be an unusual outcome of the aberrant DNA repair process in cancer cells under stress in which repair may be guided more towards fixing fatal DNA damage, for example, truncation of BCR-ABL, to avoid cell death, whereas it occurs with compromised repair fidelity,89,90 leading to nonfatal point mutations. This study may have broader implications for the biology of cancer evolution. As cancer cells tend to produce higher amounts of reactive oxygen species and have higher levels of endogenous DNA damage, 91 increased error-prone DNA damage repair by the stress response gene SIRT1 in cancer cells may provide a pathway to facilitate the accumulation of genetic mutations and accelerate cancer evolution under endogenous oxidative stress. In this regard, cancer evolution under stress may resemble the adaptive mutagenesis process of bacteria under stressful conditions.80,92 More studies are needed to further uncover the precise molecular mechanisms of SIRT1 in therapeutic and endogenous stress signaling and DNA repair. Nonetheless, the study by Wang et al. 88 provides the first evidence that acquired resistance through mutation acquisition can be regulated by SIRT1. Acquired resistance is a widespread phenomenon in targeted therapy, and it would be interesting to determine in the future if acquired resistance through acquisition of mutations in other types of cancer can be regulated and how SIRT1 may play a role in those settings.
SIRT1 Facilitates Gain of Function of Cancer Stem Cell Properties
Cancer stem cells (CSCs), also known as “tumor-initiating cells,” are the cancer cell population that carries functional properties of stem cells: self-renewal, the capability to differentiate into multiple lineages in the tumor overall, and the potential to proliferate extensively to expand the malignant cell population. 93 CSCs have been identified in hematological malignancies, breast cancer, and brain tumors. 93 CSCs may arise from self-renewing stem cells by the acquisition of mutations or from more differentiated cells after the gain of function of CSC properties.94,95 CSCs are essential for cancer development and can serve as the source of primary cancer or a reservoir of cells with the capability of relapse or metastasis. In chronic phase CML, imatinib fails to eradicate CML stem cells,96,97 and residual leukemic stem cells may eventually cause disease relapse after imatinib cessation. SIRT1 expression level is shown to be higher in CSCs than in differentiated tumor cells, for example, human CD133+ glioblastoma stem cells, 98 and human and murine CML stem cells.26,54 Yuan et al. 26 showed that SIRT1 knockout or inhibition hinders BCR-ABL transformation of hematopoietic stem cells and CML disease development, providing direct evidence of a crucial role of SIRT1 in CSCs and leukemogenesis.
CSCs possess multiple mechanisms of drug resistance: highly expressed ABC transporter, highly expressed antiapoptosis factors, and quiescent status to avoid replication stress and apoptosis induction. Based on the above discussion, it is not surprising that SIRT1 plays a critical role in the drug resistance of CSCs. Li et al. 54 showed that SIRT1 inhibition increases p53 acetylation, sensitizes CML stem cells to imatinib treatment, and enhances the elimination of CML stem cells. This study not only demonstrates an important role of SIRT1 in CSC-mediated drug resistance but also provides proof of principle that SIRT1 inhibition may be employed to eliminate residual CML disease and improve the therapeutic outcome of TKIs. In addition to CML, SIRT1 knockdown enhances radiosensitivity and radiation-induced apoptosis in glioma CD133+ progenitor cells. 99
Transiently acquired CSC properties also contribute to drug resistance during cancer therapy. Sharma et al. 100 showed that treatment with epidermal growth factor receptor TKI gefitinib induces a subpopulation of non–small cell lung cancer cells to transiently acquire CSC properties by chromatin modification and thus resist TKI treatment. Such a mechanism may contribute to cancer acquired resistance without acquisition of genetic mutations, and it may be prevented by epigenetic approaches such as HDAC inhibitors. 100 Transient acquisition of CSC properties is also observed during cancer epithelial-to-mesenchymal transition (EMT). EMT promotes the acquisition of invasive and metastatic properties of cancer cells. With transcriptional reprogramming during the EMT process, cancer cells could change their phenotypic display as a result of partial dedifferentiation and acquire CSC properties, resulting in drug resistance. 101 SIRT1 is found to be a positive regulator of EMT. 102 Overexpression of SIRT1 induces EMT through transcription factor ZEB1 in prostate cancer cells; SIRT1 knockdown restores cell-cell adhesion and reverses the EMT of prostate cancer cells in vitro and in vivo. Given that SIRT1 is an epigenome regulator, SIRT1 may play an important role in mediating dedifferentiation and facilitating the acquisition of CSC properties of cancer cells for drug resistance. More studies are anticipated in this area.
SIRT1 Alters the Tumor Microenvironment
Rapid tumor growth creates a hypoxic microenvironment characterized by a disorganized vascular architecture and irregular blood flow. The tumor microenvironment not only limits the effective delivery of anticancer drugs for innate resistance, as discussed above, 3 but also promotes acquired resistance through cell-nonautonomous effect. Several recent studies have shown that tumor microenvironment–secreted factors, hepatocyte growth factor (HGF) and the Wnt family member 16B (WNT16B), provide strong survival signals in a paracrine manner to cancer cells for innate and acquired resistance.103-105 In addition, an altered microenvironment provides a combination of cytokines that nurture and facilitate the survival of CML leukemic stem cells, which also contributes to drug resistance. 106
Although direct roles of SIRT1 in tumor microenvironment–mediated drug resistance have not been shown, several lines of evidence have indicated that SIRT1 is involved in the process forming the tumor microenvironment. Pathological angiogenesis is a hallmark of cancer that is critical for tumor growth and metastasis. 107 Angiogenesis is controlled by the balance of proangiogenic and antiangiogenic molecules. It has been shown that SIRT1 controls endothelial angiogenic functions through deacetylating FOXO1 and Notch1 intracellular domain (NICD) during vascular growth.108,109 SIRT1 enhances tumor angiogenesis through negatively modulating Delta-like ligand 4 (DLL4)/Notch signaling in Lewis lung carcinoma xenograft-derived vascular endothelial cells. 110 SIRT1 also activates endothelial nitric oxide synthase by deacetylation to enhance nitric oxide production and improve vascular function. 111 As a consequence, enhanced tumor angiogenesis induced by SIRT1 may bring more nutrition to cancer cells and facilitate their survival and growth. In addition, it has been reported that SIRT1 helps cells survive against a hypoxic environment by activating hypoxia-inducible transcription factors HIF2α while contrastingly inactivating HIF1α.112,113 More studies are needed to further clarify and understand how SIRT1 regulates the tumor microenvironment for drug resistance.
The Potential Undesired Impact of SIRT1 Inhibition
At this point, SIRT1 inhibition appears to be a good approach to overcome cancer drug resistance and improve cancer therapy. However, caution should be exercised because SIRT1 might have tumor suppressor functions, and prolonged SIRT1 inhibition could have adverse effects. The tumor suppressor role of SIRT1 is illustrated in several mouse studies demonstrating that SIRT1 overexpression reduces colon and intestinal polyps in APCmin/+ mice, 114 thymic lymphoma in p53+/− mice, 59 and spontaneous carcinoma/sarcoma or carcinogen-induced carcinoma. 115 In addition, heterozygous loss of SIRT1 accelerates cancer development in the p53+/− genetic background. 43 It is believed that SIRT1 may exert its antitumor functions by improving genome stability through enhancing DNA damage repair.43,59 Alternatively, SIRT1 may help protect mice from tumorigenesis in a cell-nonautonomous manner via improving whole body metabolism. It is known that SIRT1 enhances gluconeogenesis and increases glucose output through modulating FOXO1 and PGC-1α in the liver, 116 regulates adipogenesis and lipid metabolism through PPARγ and LXR,47,117 and increases insulin secretion and sensitivity.118,119 Overexpression of SIRT1 in mice improves glucose tolerance under a high-fat diet and protects animals from metabolic damage.120,121 In addition, SIRT1 regulates circadian clock gene expression to control metabolic rhythm.122,123 Considering that cancer is a systematic disease related to disrupted metabolism, SIRT1 may serve as a guard against cancer by providing tight regulation of metabolism in the proper amplitude. 124
Prospect of SIRT1 Inhibition for Cancer Treatment
Given the above consideration, it is important to know that SIRT1 inhibition may not necessarily increase cancer incidence in mice. At least 3 published works showed that SIRT1−/− mice from different genetic backgrounds that underwent aging studies do not die of obvious cancer.125-127 Similarly, in our hands, aging SIRT1−/− mice did not show an increased tumor incidence (unpublished data). As a matter of fact, SIRT1 knockout moderately reduces the intestinal polyp number and surface in Apcmin/+ mice. 128 Most strikingly, SIRT1 knockout significantly inhibits BCR-ABL transformation of hematopoietic stem cells and leukemia development, 26 clearly suggesting an oncogenic effect of SIRT1 on bone marrow stem cells. Furthermore, Herranz et al. 129 showed that SIRT1 overexpression in transgenic mice inhibits prostate and thyroid cancers induced by the loss of tumor suppressor PTEN. Together, mouse studies support that SIRT1 can be either a tumor suppressor gene or an oncogene, depending on the cellular context. Activation or inhibition of SIRT1 may be used to serve different biological purposes. Controlled SIRT1 inhibition may provide a novel strategy to overcome cancer drug resistance.
Numerous SIRT1 small molecule inhibitors have been developed or are under development. 41 Among them, SIRT1 inhibitors tenovin-620,26,54,130 and cambinol 67 have been shown encouraging an in vivo effect against cancers. However, these inhibitors are neither potent enough nor specific enough, and the in vivo effect is also limited. 26 SIRT1 activity is dependent on the balance of endogenous levels of NAD+ and NAM. Strategies to modulate cellular NAD+ and NAM levels can also be used to regulate SIRT1 activity. However, such approaches may affect other sirtuins and NAD+-dependent enzymes. Therefore, more potent and specific SIRT1 inhibitors need to be developed for human clinical studies. The new generation of SIRT1 inhibitors in combination with other specific anticancer agents may have promising effects on overcoming cancer drug resistance and improve therapeutic outcomes of cancer treatment.
Footnotes
Acknowledgements
The authors acknowledge research support from the National Cancer Institute of the National Institutes of Health (R01 CA143421) and the State of California Tobacco-Related Disease Research Program (20XT-0121) to W.Y.C. The contents are solely the responsibility of the authors and do not represent the official views of the National Institutes of Health.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: Research support from the National Cancer Institute of the National Institutes of Health (R01 CA143421) and the State of California Tobacco-Related Disease Research Program (20XT-0121) to W.Y.C.