
Abstract
Cell cycle deregulation is a common motif in human cancer, and multiple therapeutic strategies are aimed to prevent tumor cell proliferation. Whereas most current therapies are designed to arrest cell cycle progression either in G1/S or in mitosis, new proposals include targeting the intrinsic chromosomal instability (CIN, an increased rate of gain or losses of chromosomes during cell division) or aneuploidy (a genomic composition that differs from diploid) that many tumor cells display. Why tumors cells are chromosomally unstable or aneuploid and what are the consequences of these alterations are not completely clear at present. Several mitotic regulators are overexpressed as a consequence of oncogenic alterations, and they are likely to alter the proper regulation of chromosome segregation in cancer cells. In this review, we propose the relevance of TPX2, a mitotic regulator involved in the formation of the mitotic spindle, in oncogene-induced mitotic stress. This protein, as well as its partner Aurora-A, is frequently overexpressed in human cancer, and its deregulation may participate not only in chromosome numeric aberrations but also in other forms of genomic instability in cancer cells.
TPX2 as a Microtubule Regulator
TPX2 was first described in 1997 when Heidebrecht and colleagues
1
detected a 100 kDa protein whose expression was induced from the G1/S transition to cytokinesis. TPX2 was then reported to localize to the nucleus during S-phase and G2 and at the mitotic spindle poles during mitosis (Fig. 1, A, and B). In the following decade, different studies described a critical role for TPX2 in spindle formation and dynamics. Two papers in 1998 and 2000 showed that TPX2 was critical for spindle pole organization and the localization of the kinesin-like protein Xklp2 in the Xenopus laevis egg mitotic spindle,2,3 thus originating the current name of the protein (
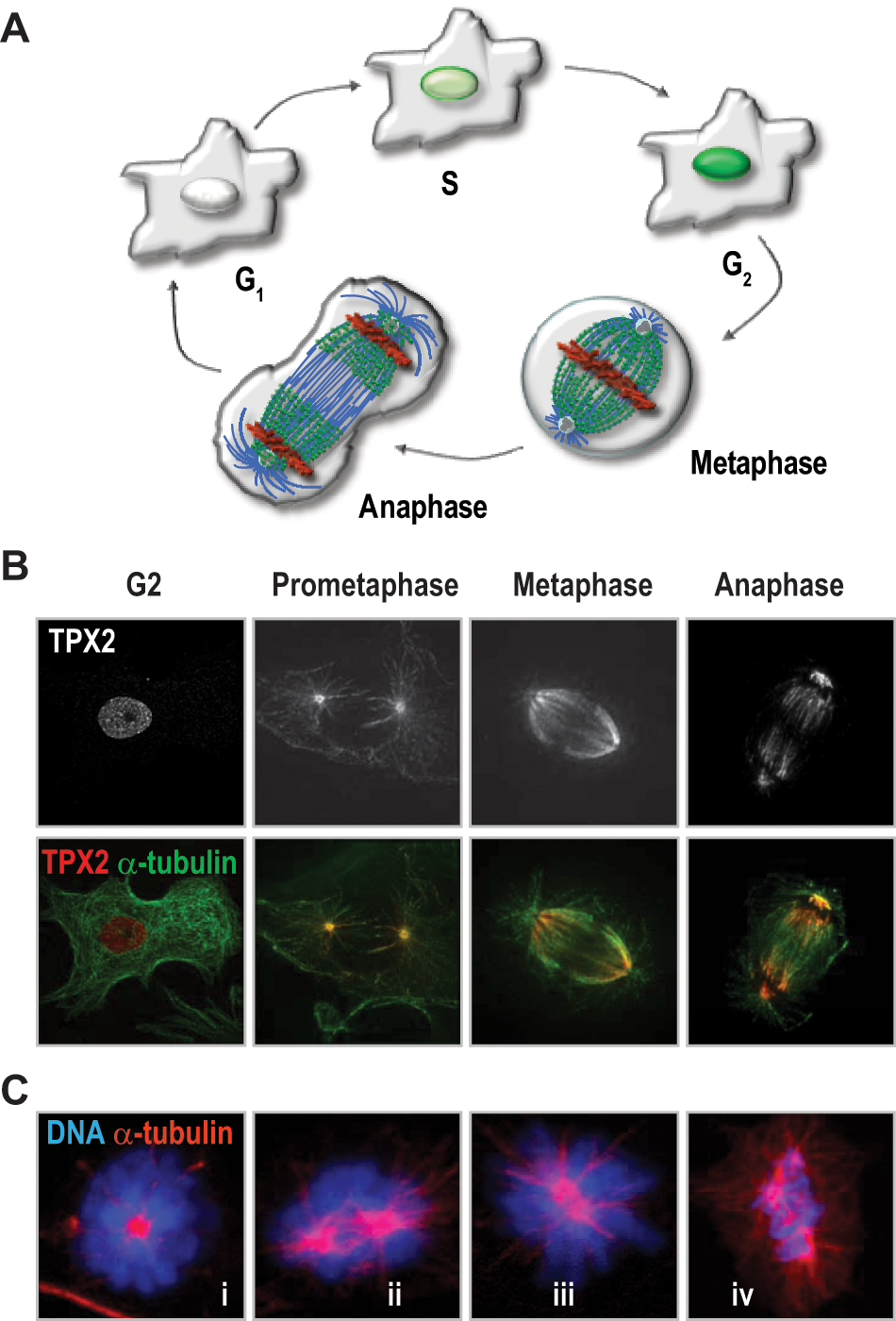
TPX2 in the mammalian cell cycle.
In addition to its critical role in spindle formation, TPX2 is thought to participate in other processes during mitosis or meiosis. TPX2 is involved in the reassembly of the nucleus of the daughter cells after mitosis in Xenopus egg extracts. 10 In neural progenitors, TPX2 is involved in cell-cycle-dependent interkinetic nuclear migration, a process in which microtubules are necessary for apical movements of the nucleus after DNA replication. 11 More recently, a novel function as a scaffold and co- activator protein of the chromosome passenger complex (CPC) has been proposed. 12 TPX2 interacts with the main members of the CPC, Aurora-B, Survivin, and INCENP in Xenopus egg extracts and 293T cells. Endogenous TPX2 may increase Aurora B activity, whereas TPX2 overexpression could inhibit the activity of this kinase, although further experiments are necessary to validate the relevance of these data. TPX2 has been shown also to govern cell cycle progression in mouse oocytes, suggesting specific functions during meiosis. 13 TPX2 accumulates from meiosis I to meiosis II and is required for spindle assembly in oocytes. In these cells, TPX2 controls microtubule assembly and spindle stability throughout the phosphorylation of Aurora-A-dependent phosphorylation of TACC3, a regulator of microtubule organizing centers.13-15
TPX2 Structure and Regulation
The primary sequence of the TPX2 protein is relatively well conserved through evolution, although a TPX2 orthologue is not present in yeast, and some specific domains are not evident in lower animals16,17 (Fig. 2A). This conservation is also maintained at the functional level since TPX2 plays similar roles in different organisms such as plants, ascidians, worms, flies, and vertebrates.16-19 The TPX2 protein presents 2 main functional domains and several regulatory sequences (Fig. 2B). The N-terminal end is involved in binding to Aurora-A,20,21 whereas the C-terminal domain is responsible for binding to microtubules. 22 The very last 35 amino acids in the C-terminal region are necessary for binding to the class 5 kinesin-like motor protein EG5. 23 A short central sequence in TPX2 is involved in importin-α binding, and along the nuclear localization signals (NLS) these regions are important for regulation of subcellular localization during the cell cycle. 24 The 43-aa domain at the N-terminus is sufficient for activation of Aurora-A. This domain does not provoke global conformational changes in the kinase, but a helical fragment of TPX2 (residues 30-43; Fig. 2C) binds Aurora-A near helix αC. The activation segment is constrained in an inward conformation that is primed for substrate binding and that protects a critical activating residue of Aurora-A from dephosphorylation by PP1. 20
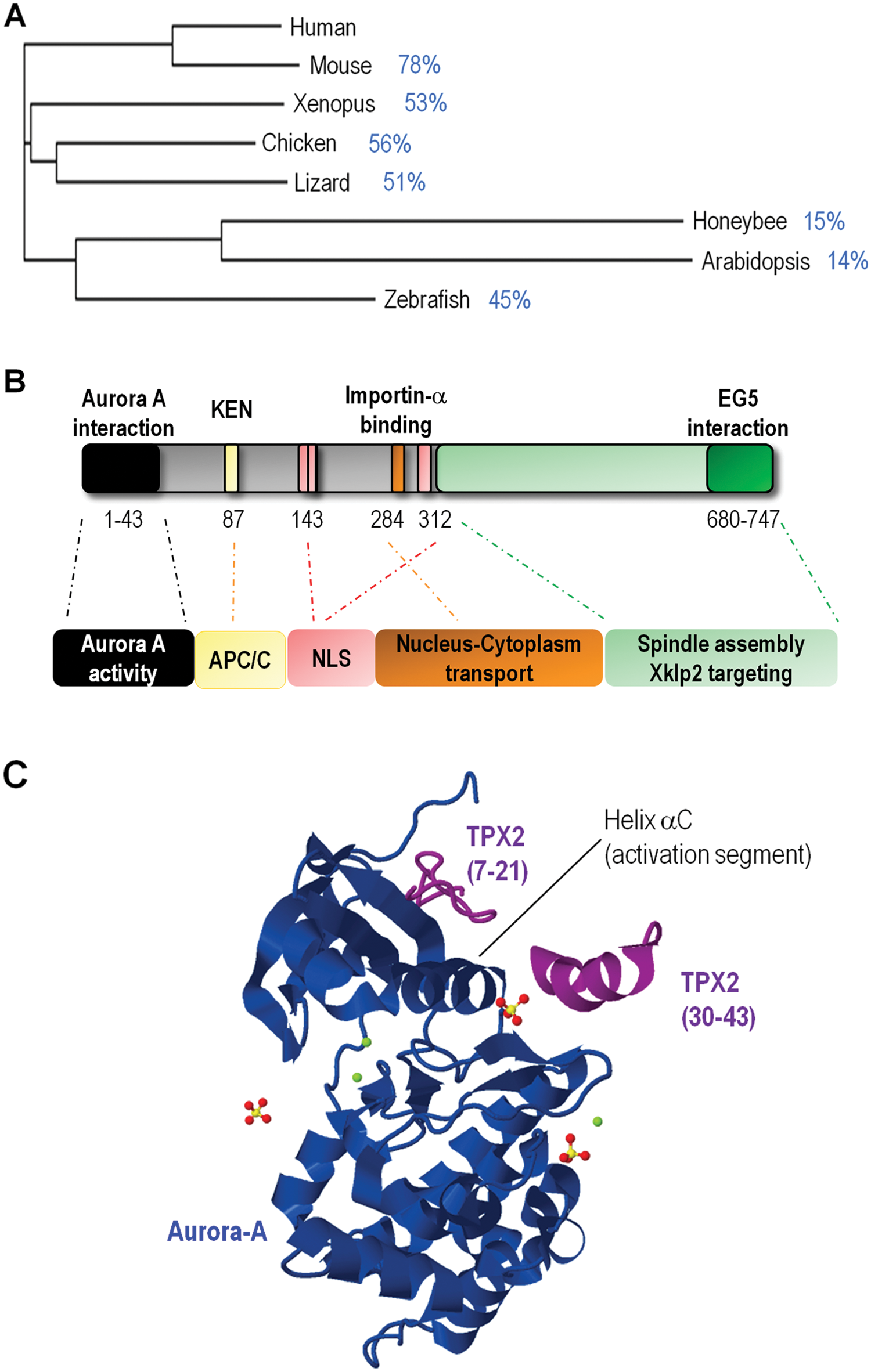
TPX2 structure and regulation.
No detailed information has been reported about the transcriptional regulation of TPX2. The fact that this protein is detected in proliferating cells from S to mitosis is compatible with an E2F-dependent regulation of the TPX2 gene, which in fact contains several E2F consensus sites in its promoter region and binds E2F transcriptional factors in several cell types. 25 The fact that TPX2 is also expressed in postmitotic neurons (see below) suggests additional mechanisms of transcriptional regulation independent of proliferation. One of the most interesting aspects of the synthesis of TPX2 expression is the fact that this protein can be directly translated at the spindle. 26 During meiotic progression, multiple maternal mRNAs must be translated. This process is regulated by the specific elements that are present in their 3′UTRs. Among those elements, the cytoplasmic polyadenylation element (CPE) recruits the CPE-binding protein CPEB that dictates the timing and extent of translational activation. 27 TPX2 is one of the genes whose translation is regulated by this mechanism during meiosis. More importantly, its translation occurs directly at the sites where TPX2 plays a more critical function, the spindle. It is interesting to point out that the localized translation of not only TPX2 but also other mitotic regulators containing CPEs (including BUB1, EG5, MAD2, and Aurora-A) is required for chromosome segregation and meiotic progression in these cells. 27
Finally, the levels of TPX2 are tightly regulated during mitotic exit to prevent its activity during the following cell cycle. This is achieved by ubiquitin-dependent proteasome-dependent degradation. The E3-ubiquitin ligase APC/C (anaphase-promoting complex/cyclosome), bound to its coactivator CDH1, is responsible for targeting TPX2 with ubiquitin and triggering the proteasome-dependent degradation of this protein during anaphase-telophase and the following G1 phase. 28 Genetic ablation of CDH1 results in a significant overexpression of TPX2, although this does not have dramatic consequences, at least during mitotic exit. 29
The relevance of the posttranslational modifications of TPX2 is not well understood. Human and Xenopus TPX2 are phosphorylated during mitosis.1,3 This modification may depend on Aurora-A kinase activity, although the mutation of the 3 putative serines phosphorylated by Aurora-A does not affect the activation of the TPX2 kinase partner.30,31 Plx1, the Xenopus orthologue of PLK1, phosphorylates TPX2 at Ser204, and this seems to increase Aurora-A activity. 32 Yet, the functional relevance of these modifications is not well established at present.
The TPX2-Aurora-A holoenzyme
TPX2 was identified by mass spectrometry as a major partner of the kinase Aurora-A in mitotic HeLa cell extracts. 31 Aurora-A is a key regulator of mitosis involved in centrosome maturation and separation, mitotic entry, and spindle assembly [reviewed in Carmena et al. 33 ]. In the mouse, lack of Aurora-A, similar to TPX2 ablation, results in embryonic lethality at the morula-blastocyst transition due to defects in mitosis.34-36 During the cell cycle, Aurora A and TPX2 have similar expression patterns characterized by a progressive increase of expression from S/G2 to mitosis. Despite the coincidence in expression, the subcellular localizations of Aurora-A and TPX2 are not identical, at least during interphase. Aurora-A is present on duplicated centrosomes from late S phase until early G1 phase, whereas interphase TPX2 is mostly nuclear. During mitosis, both proteins co-localize at the spindle apparatus, although Aurora-A maintains a preference for spindle poles and TPX2 is more abundant in the spindle itself. 31 At the end of mitosis, both proteins are targeted for degradation by the APC/C E3-ubiquitin ligase.28,37,38
The mechanisms of activation of Aurora-A are still under deep investigation, although it is clear that TPX2 is a major regulator of its activity at different levels. TPX2 is involved in the localization of Aurora-A at the spindle,17,31 stabilization of Aurora-A protein levels, 39 and achievement of appropriate levels of kinase activity.20,21,40 The TPX2-Aurora-A binding occurs between the catalytic C-terminal part of the kinase and the N-terminal part of TPX2 (Fig. 2, B and C). This binding is required for a proper localization of Aurora-A since spindle defects found in TPX2-deficient cells are accompanied by a significant reduction in the microtubule associated localization of this kinase as well as the kinesin KIF15 (mammalian homologue of Xklp9,31,41,42). Aurora-A participates in spindle formation by regulating different microtubule associated proteins including the microtubule bundling factor HURP, the microtubule stabilizer XMAP215, and the microtubule destabilizing protein MCAK.43-46 TPX2 can also regulate Aurora-A stability by inhibiting the APC/C-dependent degradation of this kinase during G2 and mitosis. 39 Whether this is a consequence of the role of TPX2 in the proper localization of Aurora-A is not clear.
Aurora-A is activated through the autophosphorylation of a threonine residue located in the catalytic domain and inhibited by the dephosphorylation of this residue by the phosphatase PP1. Different Aurora-A cofactors facilitate this autophosphorylation including BORA, AJUBA, and PAK1 in addition to TPX2.20,21 Binding of TPX2 to the mitotic kinase changes its 3 dimensional structure in such a way that (1) it facilitates autophosphorylation and (2) it shields the phosphorylated threonine from dephosphorylation by PP1 (Fig. 2C).20,21,40 This activating mechanism is RAN-dependent since RAN-GTP stimulates the TPX2-Aurora A interaction and, in turn, promotes the activation of Aurora-A at the microtubules. 47 The binding of TPX2 to Aurora-A is also thought to increase the binding affinity of both ATP and substrates and may decrease the accessibility to inhibitors. 48
TPX2 in Chromosomal Instability and Human Cancer
Expression of TPX2 in human cancer
TPX2 was originally described as the antigen of Ki-S2, a mouse monoclonal antibody raised in an effort to find markers of proliferation with diagnostic and prognosis value. 1 That antigen was later renamed by the same group as repp86 (restrictedly expressed proliferation-associated protein 49 ). The fact that TPX2/repp86 could serve as a proliferative marker in tumor samples drove different groups to study the expression of this protein in human cancer (see below). In addition, human TPX2 gene is located at 20q11, 50 a chromosome region frequently amplified in cancer. 51 DNA copy number increase at 20q has been found associated with the overexpression of TPX2 in different tumor types including cervical, lung, bone, and ovarian cancers.52-55 TPX2 is frequently overexpressed in many other tumor types without evidences for DNA amplification, similar to many other mitotic regulators56,57 (Fig. 3). Using available expression databases, Asteriti et al. 57 reported that up to 27% of all comparative analyses of tumor versus normal tissues were characterized for a significant overexpression of TPX2. TPX2 overexpression affects a wide range of different tumor types including astrocytomas, oral squamous cell carcinoma, bone, lung, colon, liver, cervical, salivary gland, mesothelial, ovarian, and pancreatic cancers (Table 1). Importantly, TPX2 expression is a marker of worse tumor prognosis in several malignancies including brain, breast, colorectal, laryngeal, and lung cancers, as well as meningiomas, mantel cell lymphomas, and mesotheliomas (Table 1). TPX2 expression may be also associated with tumor metastasis. A network analysis performed with a total of 5 data sets from 2 different species (human and mouse) identified a number of networks that were significantly associated with breast tumor progression. 58 Specifically, TPX2 was included together with other 8 mitotic regulators (BUB1, UBE2C, CDC20, CCNB2, KIF2C, BUB1B, CEP55, CENPA) in a tumor-cell autonomous network associated with metastatic progression.
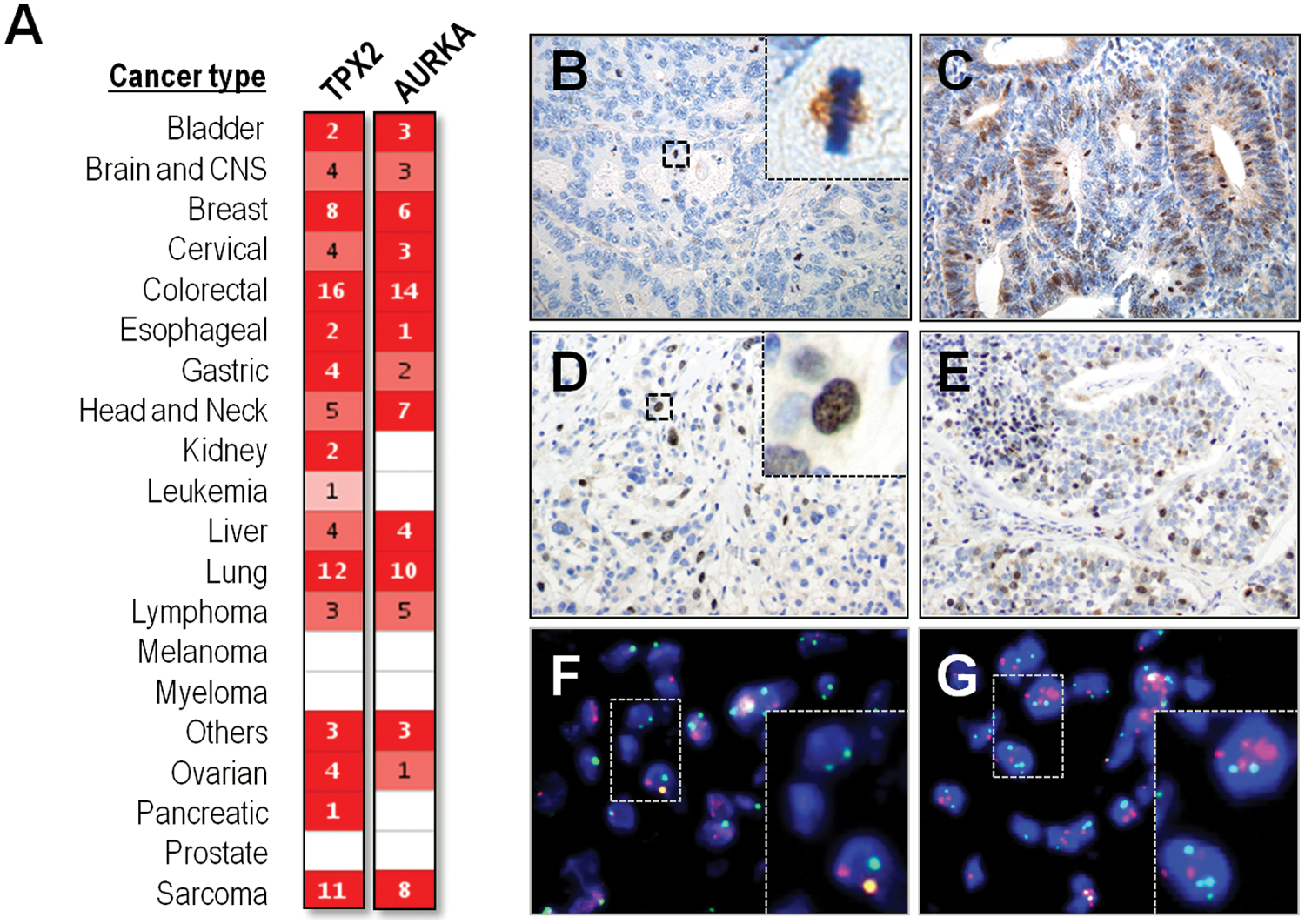
Overexpression of TPX2 and Aurora-A in human tumors.
TPX2 Overexpression in Human Cancers
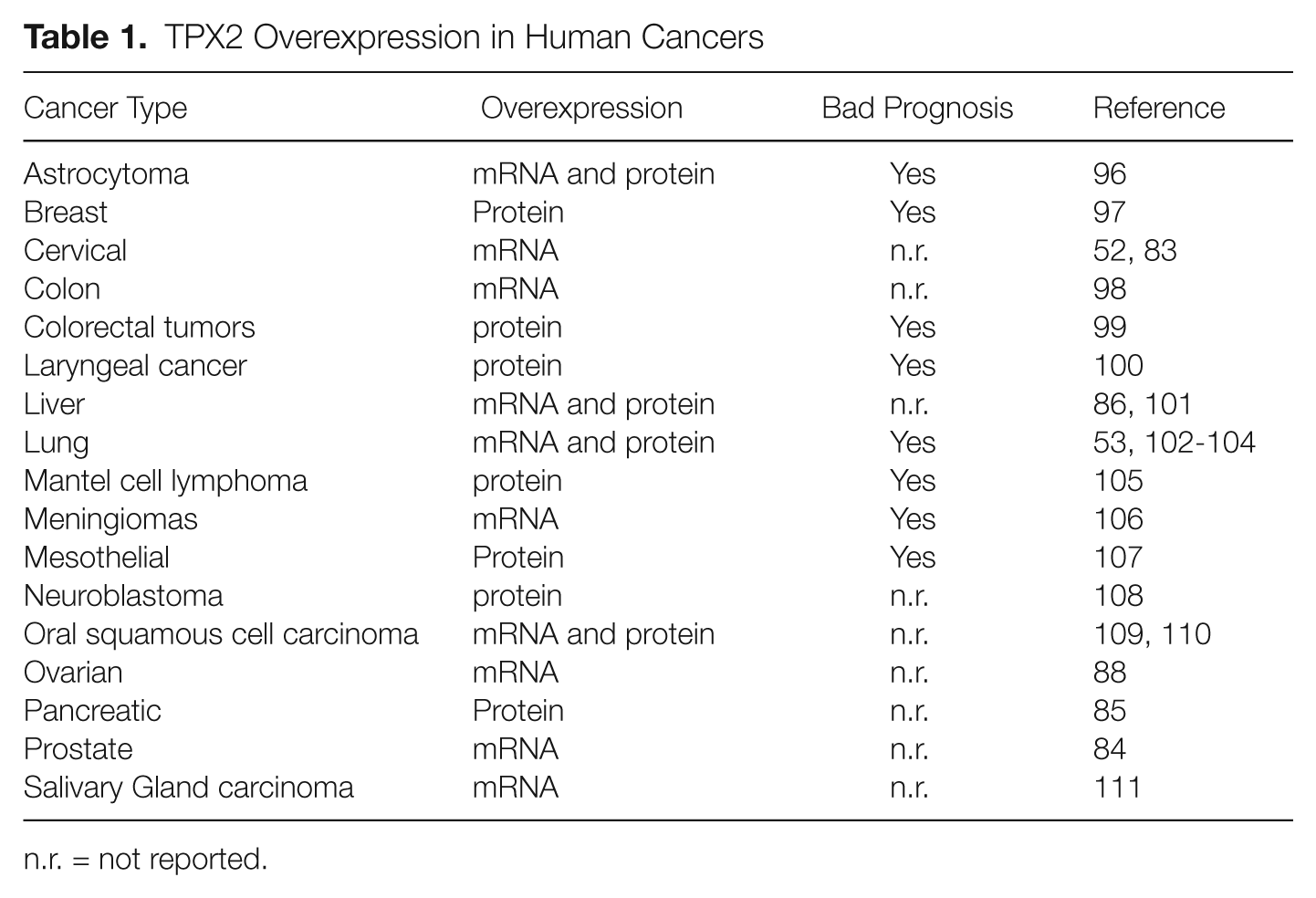
n.r. = not reported.
Although the presence of specific small mutations has not been especially studied for TPX2, a number of tumor-associated mutations are found in the public repositories indicating some concentration of TPX2 mutations in tumors in the gastrointestinal tract (COSMIC database; http://cancer.sanger.ac.uk/cosmic/). Some of these mutations result in nonsense substitutions suggesting loss of function, although their functional relevance has not been evaluated so far. Similarly, a mutation in Aurora-A that prevents binding to TPX2 is known to be oncogenic, 59 suggesting that loss-of-function alterations in either TPX2 or Aurora-A may also be present in human tumors. Altogether, published data indicate that deregulation of TPX2 is a frequent feature of tumor cells and is significantly associated with the more advanced stages of human cancers of diverse origin.
TPX2 overexpression correlates with chromosomal instability
It was a century ago when an association between cancer and abnormal chromosome segregation was suggested for the first time. 60 Although still controversial, many authors have proposed that chromosomal instability (CIN) and the subsequent aneuploidy can promote tumor development.61,62 Tumor cells may acquire CIN due to defects in the mitotic checkpoint (spindle assembly checkpoint or SAC) that prevents defective chromosome segregation.56,63 Other defects such as abnormal centrosome number64-66 of faulty chromatid cohesion may also be involved. 67 CIN and aneuploidy may have adverse effects in proliferating cells 68 but may eventually lead to the selection of abnormal genomic compositions in which the levels of specific oncogenes or tumor suppressors are altered. As an example, CIN has been show to favor loss of heterozygosity in the p53 locus leading to defective tumor suppressor activity.69,70
Cells display multiple tumor suppressor pathways in response to oncogenic signals. These include oncogene-induced senescence (OIS) and oncogene-induced replicative stress (OIRS). During OIS, the hyperactivation of mitogenic pathways results in the activation of the pRb (retinoblastoma protein) and/or p53 pathways that prevent cell cycle progression and is accompanied by other features that result in OIS and an irreversible cell cycle arrest. 71 In other cases, the oncogenic signals are accompanied by defective tumor suppressor pathways (e.g., inactivation of retinoblastoma), and the mitogenic signals provoke a premature or deregulated entry into S-phase that induces replicative stress (OIRS). 72 This is in part mediated by hyperactivation of E2F transcription factors and excessive synthesis of proteins involved in the G1/S transition or the control of replication. The OIRS generated in these conditions then triggers the activation of a DNA damage–like response that prevents further cell cycle progression.
We have recently proposed the presence of an oncogene-induced mitotic stress (OIMS) response that may alter the proper regulation of mitosis in these conditions. 73 This is based on the fact that multiple mitotic regulators are E2F targets that are induced by oncogenic events such as the activation of oncogenes or inactivation of tumor suppressor proteins. Loss of pRB or p53, for instance, results in increased levels of the SAC protein MAD2. 74 As a consequence, MAD2 is frequently overexpressed in tumor cells, possibly resulting in deregulated SAC and CIN (Fig. 4). Many other mitotic regulators are co-overexpressed with MAD2 in CIN tumors. Carter et al. 75 reported a signature of 70 genes deregulated in chromosome unstable tumors. Seven of the first 10 genes with the strongest correlation with instability regulate mitosis (TPX2, PRC1, FOXM1, CDK1, TOP2A, PCNA, and UBE2C), and 33 (47%; including Aurora-A) of the 70 genes in the complete signature regulate mitosis to some extent. Of all these genes, TPX2 displays the highest correlation with chromosomal instability. 75 Cancers such as breast cancer, and anaplastic thyroid carcinoma are among the tumor types that frequently show TPX2 overexpression and aneuploid phenotypes (Fig. 3).
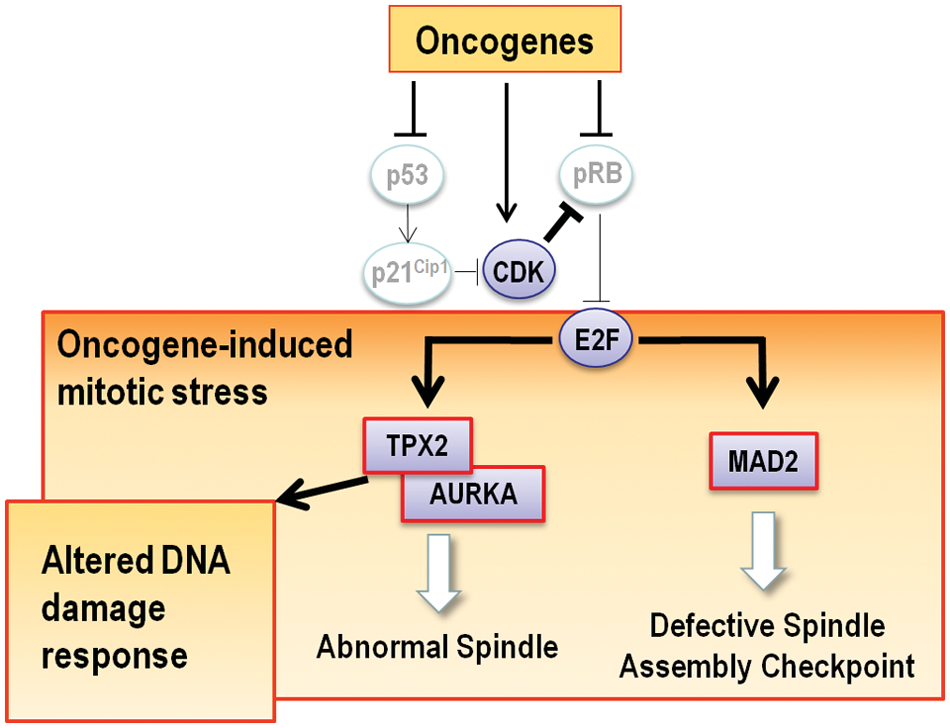
Oncogene-induced mitotic stress and TPX2. The activation of oncogenes or loss of tumor suppressor such as pRB or p53 frequently results in the overexpression of E2F transcriptional targets such as MAD2 and possibly TPX2. We propose here that overexpression of TPX2 is likely to result in abnormal Aurora-A activity and deregulated spindle dynamics. These defects occur in the presence of abnormally high levels of MAD2, known to disrupt the normal regulation of the spindle assembly checkpoint. These alterations are possibly involved in the chromosomal instability observed in tumors that overexpress these mitotic regulators. Since TPX2 and Aurora-A may play distinct roles in the DNA damage checkpoint or in the recovery from this checkpoint (see main text), its overexpression may also disturb the regulation of the DNA damage response or the efficacy of specific drugs used in chemotherapy.
The involvement of some overexpressed genes such as HEC1 or MAD2 in CIN is likely to be associated with their essential role as regulators of microtubule-kinetochore attachments and the SAC. But what is the contribution of other overexpressed proteins such as TPX2? Although it has not been analyzed in detail, overexpression of TPX2, and the probable hyperactivation of Aurora-A usually co-expressed in these tumors, are likely to disrupt the normal dynamics of the mitotic spindle, leading to defective chromosome segregation (Fig. 4). Yet the evidence for this proposal mostly comes from loss-of-function studies and not from gain-of-function models. During the last few years, different groups have demonstrated that lack of 1 of the 2 alleles of some mitotic regulators provokes a cancer-prone phenotype. This is the case for the murine MAD2, 76 Aurora-A, 36 and TPX2. 9 TPX2 is haploinsufficient to maintain genomic instability, as TPX2-heterozygous mice develop tumors in significantly higher ratio that their wild-type counterparts. 9 As expected, these tumors are highly aneuploidy, suggesting a causal connection between deregulated expression of TPX2, aneuploidy, and cancer. As demonstrated for other models such as MAD2,76,77 it is likely that the overexpression of TPX2 may result in similar defects in spindle dynamics and chromosome segregation, although this has not been properly addressed in vivo. The concomitant overexpression of MAD2 in the same tumors is likely to disrupt a normal function of the SAC, thus facilitating CIN in these conditions (Fig. 4).
Future Perspectives
Aurora-A-independent or nonmitotic roles of TPX2
Despite the central function of TPX2 in the activation of Aurora-A and formation of the mitotic spindle, some evidence calls for additional Aurora-A-independent or nonmitotic functions of TPX2. Conditional genetic ablation of TPX2, but not Aurora A, results in a significant induction of apoptosis in primary fibroblasts. 9 Whereas monopolar spindles are rare in TPX2-null cells, these figures are frequently observed in Aurora A–deficient fibroblasts,34-36 suggesting that this kinase may also play a role in centrosome maturation and separation in a TPX2-independent manner. An Aurora-A–independent function of TPX2 is required to bipolarize spindles since a TPX2 mutant that does not bind to Aurora-A is able to form bipolar spindles. 8 TPX2, but not Aurora-A, is thought to be necessary for the noncentrosomal microtubule assembly that assists in the peripheral redistribution of nuclear fragments in apoptotic cells. 78 Finally, TPX2 may play specific and previously unexpected roles in the DNA damage response.79,80 Upon ionizing radiation, TPX2 accumulates at DNA double strand breaks and associates with 2 regulators of the DNA damage response, MDC1 and ATM. 81 Overexpression of TPX2 results in decreased ionizing radiation-dependent γ-H2AX levels, whereas the opposite phenotype is observed upon TPX2 knockdown by RNA interference. This function is not associated with the mitotic roles of TPX2, since it is also observed in postmitotic neurons. 81 Whether Aurora-A is involved in this phenotype remains unknown. These data suggest that deregulation of TPX2 may be involved not only in chromosomal instability but also in genomic instability mediated by abnormal DNA damage response (Fig. 4).
It is also worth noting that TPX2 may have critical roles in postmitotic cells such as neurons. Both TPX2 and Aurora-A are expressed in postmitotic neurons, and TPX2 facilitates activation of Aurora-A at the neurite hillock resulting in phosphorylation of NDEL1 and recruitment of this protein to microtubules. Suppression of TPX2, Aurora-A, or NDEL1 results in severe impairment of neurite extension, suggesting a critical role for this pathway in microtubule remodeling in neurites. 82 Although the physiological relevance of some of these observations is not well established, these functions may have important implications in genomic instability, cancer, and, perhaps, cancer therapy.
TPX2 and cancer therapy
Because of its essential role in mitosis, elimination of TPX2 prevents proliferation of cancer cells. Target validation studies using short interfering RNAs (siRNA) in cancer cell lines have shown the potential of TPX2 in cancer therapy. Specifically, TPX2 knockdown induces cell cycle arrest, apoptosis, and the inhibition of cell proliferation and invasion in cervical, prostate, hepatocellular, and pancreatic cancer cell lines.83-86 siRNA-mediated knockdown of TPX2 inhibits the proliferation and growth of hepatocellular and pancreatic cancer cell xenografts transplanted into immunodeficient mice.85,86 Furthermore, TPX2 downregulation has been shown to sensitize pancreatic cancer cells to paclitaxel treatment. 85 Whereas the use of low doses of paclitaxel or the partial elimination of TPX2 has no significant effects on the viability of pancreatic cancer cells, the combination of these treatments reduces the viability in a dramatic manner. Interestingly, this effect is not observed when other antitumoral drugs such as gemcitabine are used, 85 and this suggests that inhibition of some components of the mitotic machinery, as also reported for Aurora-A, may synergize with microtubule poisons.87,88 These examples suggest that targeted inactivation of TPX2 may have therapeutic benefits. However, TPX2 does not display any enzymatic activity, and most current therapies are designed to inhibit its partner Aurora-A using small-molecule ATP competitors.89,90 Recently, the TPX2-Aurora-A complex has been proposed as the target of withanone, 91 a purified component of the leaf extract Ashwagadha with antitumoral properties.92,93 Treatment of cancer cells with withanone results in the dissociation of TPX2-Aurora-A complexes in an ATP-independent manner. Yet, the mechanism behind these results and their therapeutic relevance requires further studies.
It is now clear that deregulation of multiple oncogenic pathways provokes the alteration of many cell cycle regulators resulting in unscheduled proliferation as well as genomic and chromosomal instability. 94 Overexpression of TPX2 is among the most frequent markers for chromosomal instability, and this is likely linked to the critical function of this protein in microtubule dynamics and chromosome segregation. However, other new functions for this protein are now emerging, and further understanding of the functional relevance of TPX2 may facilitate the design of therapeutic approaches in the near future.
Footnotes
Acknowledgements
We thank Cristina Aguirre for her work on TPX2 function and useful discussions.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Ministerio de Economía y Competitividad (MINECO; SAF2010-19710 to I.P.C.; and SAF2012-38215 to M.M.), Fundación Ramón Areces, the OncoCycle Programme (S2010/BMD-2470) from the Comunidad de Madrid, the OncoBIO Consolider-Ingenio 2010 Programme (CSD2007-00017) from the MINECO, and the European Union Seventh Framework Programme (MitoSys project; HEALTH-F5-2010-241548).