
Abstract
The mammalian COOH-terminal binding proteins (CtBPs) CtBP1 and CtBP2 are metabolically regulated transcriptional co-repressors that are degraded upon acute exposure to the alternative reading frame (ARF) tumor suppressor. We reported previously that CtBP stimulates cell migration in certain contexts via repression of PTEN transcription and activation of the phosphatidylinositol 3-kinase (PI3K) pathway. We have now identified an additional and direct mechanism for CtBP stimulation of cell migration via regulation of T-cell lymphoma invasion and metastasis 1 (Tiam1) protein. Tiam1 is a guanine nucleotide exchange factor (GEF) for Rac GTPase that plays a critical role in regulating cell adhesion, invasion, and migration and has been directly implicated in the promotion of cancer progression and metastasis. We noted a strict positive correlation between CtBP2 and Tiam1 expression levels and that CtBP promotion of cell migration required CtBP-dependent transcriptional activation of Tiam1. RNA interference (RNAi)–mediated knockdown of CtBP2 in human colon or lung carcinoma cells led to decreased Tiam1 protein and mRNA expression, while overexpression of CtBP2 increased Tiam1 expression levels. RNAi and overexpression studies also demonstrated that Tiam1 is a key downstream mediator of CtBP2-mediated cell migration. An analysis of the Tiam1 promoter revealed binding sites for the CtBP-interacting Kruppel-like factor 8 (KLF8), and a Tiam1 promoter luciferase reporter was induced in the presence of both KLF8 and CtBP2, consistent with KLF8-dependent CtBP transactivation of Tiam1. Chromatin immunoprecipitation analyses demonstrated CtBP2 occupancy of the Tiam1 promoter that was dependent on the presence of KLF8. Our results indicate that Tiam1 is a transcriptional activation target of CtBP2 and that this interaction promotes the pro-oncogenic function of CtBP2 leading to cancer cell migration. Transcriptional activation thus plays a role in CtBP pro-oncogenic functions along with the previously characterized CtBP co-repressor function.
Introduction
COOH-terminal binding proteins (CtBPs) promote cancer cell migration, invasion, and survival via transcriptional co-repression of multiple tumor suppressor genes involved in cell motility, adhesion, and the apoptotic program.1-3 The conserved CtBP1 and CtBP2 paralogs encode a conserved central dehydrogenase homology domain with an NAD+/NADH binding region within this domain that acts as a regulatory switch, with the repressor activity induced by conformational changes caused by NADH binding.4-8 Due to the metabolic impact of hypoxia on NADH levels, CtBP has been proposed as a mechanistic link between hypoxic tumor environments and tumor progression.1,2 CtBP1 and CtBP2 are overexpressed in colon, breast, and prostate cancer, 9 and increased levels have been linked to the loss of tumor suppressors such as APC, Hipk2, and alternative reading frame (ARF). 10
CtBP2’s ability to direct cell migration has been previously linked to its ability to repress PTEN expression and thereby stimulate phosphatidylinositol 3-kinase (PI3K) activity, resulting in Rac-dependent migration. 2 The ability of the ARF tumor suppressor to inhibit migration/invasion and metastasis independent of p53 activation in animal models 11 has also been linked to its ability to inactivate CtBP through direct binding and targeting of CtBP to the proteasome. 12 Thus, a complete understanding of the mechanism by which CtBP activity is linked to the induction of cancer cell migration is critical to understanding mechanisms of ARF tumor suppression and also will likely contribute to our understanding of invasion/metastasis in human cancer.
The control of cell migration by ARF has been linked to the modulation of Rac GTPase activity, at least in part due to CtBP-mediated repression of PTEN and activation of PI3K. 2 Indeed, deregulated activity of Rac/Rho GTPases results in enhanced cancer cell migratory, invasive, and metastatic properties.13,14 In human cancers, there is increasing evidence of deregulation of GTPases such as Rac and Rho caused by deregulated expression and/or activity of associated GTP exchange factors (GEFs). 15 GEFs promote formation of the active GTP-bound state of GTPases, and GTPase-activating proteins (GAPs) return GTPases to the GDP-bound inactive state.
T-cell lymphoma invasion and metastasis 1 (Tiam1) is a GEF for Rho-like GTPases and specifically activates Rac1 in vivo and Rac, cdc42, and Rho in vitro.16-18 Tiam1 and its paralog Tiam2 are conserved among vertebrates, and Tiam1 is ubiquitously expressed in adult tissues. 19 The Tiam1-Rac1 pathway regulates numerous biological properties, especially cell adhesion and migration,20,21 and its activation is associated with an increased invasiveness of many tumors, such as nasopharyngeal carcinoma, 22 breast cancer, 23 and retinoblastoma. 24 Tiam1 expression is also transcriptionally regulated during the epithelial to mesenchymal transition. 25 Tiam1-deficient mice develop fewer and smaller Ras-induced skin tumors than wild-type mice, 26 and cells with endogenously high levels of Tiam1, or with ectopic overexpression of Tiam1, demonstrate increased cellular migration and metastatic potential in mouse models. 27 In colon tumor cells, Tiam1 affects multiple properties associated with acquisition of the metastatic phenotype and may represent a marker of colon tumor progression and metastasis in a subset of tumors. 28
In this work, we found that the CtBP-driven migration of colon cancer cells 2 was markedly accelerated by Tiam1 and that Tiam1 expression rescued migration after CtBP2 depletion. CtBP2 and Tiam1 were linked in a transcriptional pathway, where CtBP2 co-activated, not repressed, Tiam1 expression. Chromatin immunoprecipitation (ChIP) analysis demonstrated that CtBP2 was recruited to the Tiam1 promoter by the basic Kruppel-like factor (KLF8) transcription factor. Tiam1 is thus a heretofore unknown downstream activation gene target of CtBP2 required for CtBP’s ability to promote the migration of colon carcinoma cells and is likely an important downstream mediator of CtBP’s pro-oncogenic functions.
Results and Discussion
CtBP2 positively regulates Tiam1 expression
Given the critical role for Tiam1 in cellular invasion and migration 28 and the correlation of CtBP induction of migration with the activation of Rac GTPase activity, 2 we examined whether the Rac/Rho GEF Tiam1 was a direct or indirect downstream target for CtBP transcriptional regulation. HCT116 (p53−/−) cells were transfected with control or CtBP2 siRNA and then analyzed for Tiam1 protein and mRNA expression levels. Tiam1 protein abundance was reduced by approximately 40% (Suppl. Table S1) in CtBP2 siRNA-treated cells as compared with control siRNA-treated cells (Fig. 1A, left panel). To assess whether CtBP2 was regulating Tiam1 expression at the transcriptional level, Tiam1 mRNA levels were analyzed in HCT116 (p53−/−) cells 24 hours after treatment of the cells with CtBP2 siRNA (Fig. 1A, right panel). qRT-PCR analysis confirmed that Tiam1 mRNA expression was also downregulated approximately 40% (P < 0.05) upon CtBP2 depletion in HCT116 (p53−/−) cells (Fig. 1A), most likely accounting for the observed decrease in protein abundance upon CtBP2 depletion.
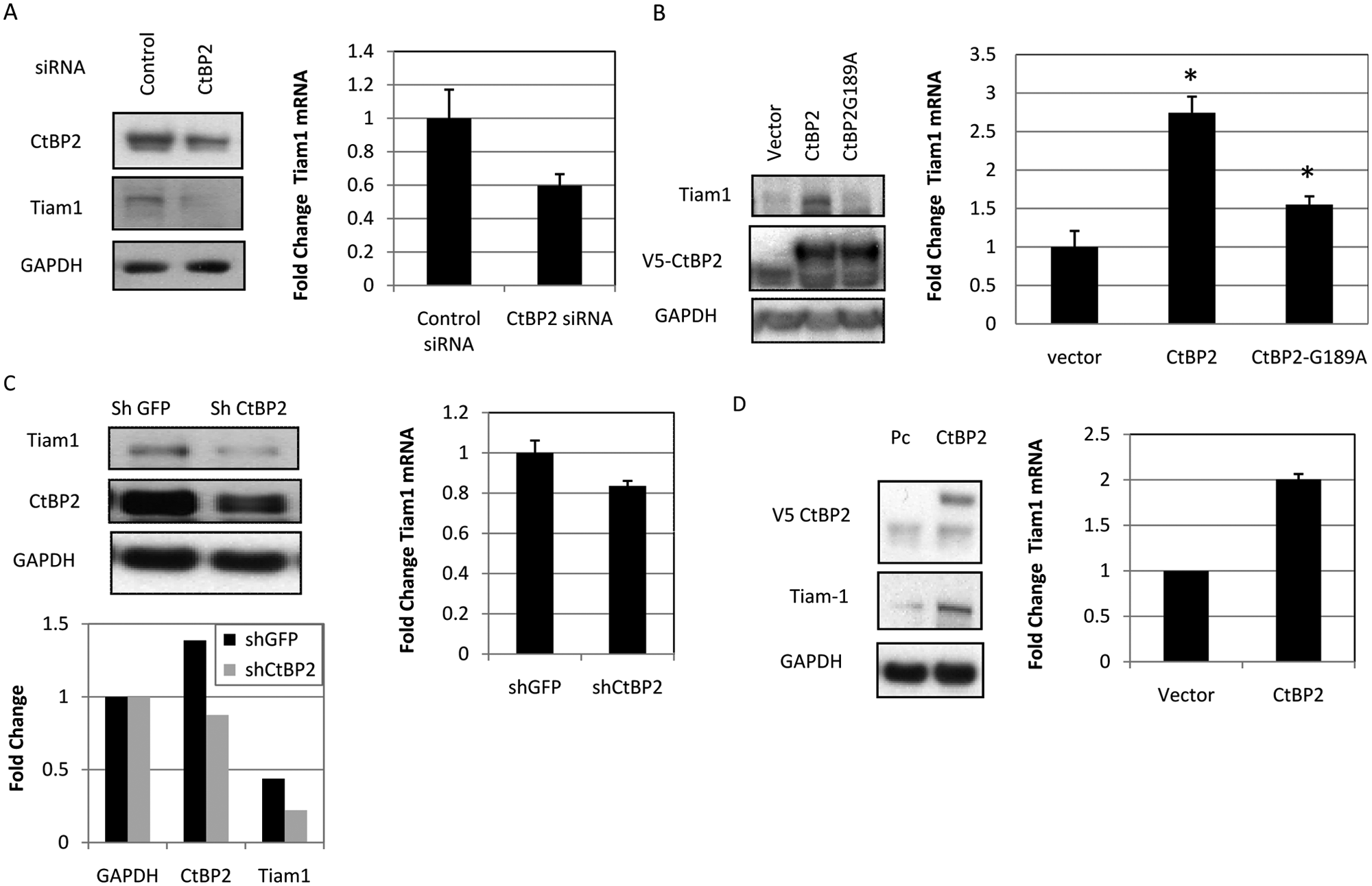
CtBP2 regulates Tiam1 protein and RNA levels in HCT116 (p53−/−) and H1299 cells. (
If CtBP2 depletion decreased Tiam1 protein and mRNA levels, then raising the level of cellular CtBP2 might be expected to increase Tiam1 protein and mRNA levels. To test the effect of CtBP2 overexpression on Tiam1 levels, control, wild-type, or G189A NADH binding mutant (transcriptional repression defective 5 ) V5-tagged CtBP2 cDNA expression vector was transfected into HCT116 (p53−/−) cells. CtBP2-expressing HCT116 (p53−/−) cells showed substantially increased (2.3-fold) Tiam1 protein levels as compared to control cells (Fig. 1B, left panel, and Suppl. Table S1). qRT-PCR also revealed a markedly increased abundance of Tiam1 mRNA after HCT116 (p53−/−) cells were transfected with CtBP2 (2.75-fold over control) (Fig. 1B, right panel), consistent with the effect of CtBP2 on Tiam1 protein levels acting at the level of mRNA abundance. Compared with wild-type CtBP2, NADH binding defective CtBP2 was significantly defective in activating Tiam1 mRNA expression (1.5- v. 2.75-fold induction; P < 0.05), with no detectable protein induction (Fig. 1B), suggesting that the effect of CtBP2 on Tiam1 mRNA levels was linked to CtBP2/NADH interaction similar to the linkage of CtBP2 transcriptional repression to NADH binding. Thus, CtBP2 appears to control Tiam1 expression through control of Tiam1 mRNA expression.
To determine if CtBP acts to increase Tiam1 mRNA and protein levels in cells other than HCT116 (p53−/−) colon cancer cells, H1299 human lung cancer cells were also tested in the same manner for CtBP2 regulation of Tiam1. Tiam1 protein and mRNA expression levels were determined by immunoblots and qRT-PCR, respectively, in H1299 cells transfected with CtBP2 shRNA. As seen in HCT116 (p53−/−) cells, CtBP2 depletion resulted in a 50% decrease in the Tiam1 protein level (Fig. 1C, top left panel and bottom left panel, and Suppl. Table S1) as well as a more modest but statistically significant (P < 0.05) 20% decrease in the Tiam1 mRNA level (Fig. 1C). Furthermore, as also seen in the colon cancer cells, CtBP2 overexpression in H1299 cells resulted in increased Tiam1 protein (3-fold) and mRNA (2-fold) levels and to a similar extent as observed in HCT116 (p53−/−) cells (Fig. 1D).
Tiam1 overexpression rescues cell migration after CtBP2 knockdown
CtBP2 expression has been previously reported to promote HCT116 cell migration, and CtBP2’s depletion completely abrogates cell migration activity. 2 To test if Tiam1 overexpression could rescue the inhibition of cell migration caused by CtBP2 knockdown, HCT116 (p53−/−) cells were first transfected with control, CtBP2 siRNA, or CtBP2 siRNA combined with Tiam1 cDNA expression vector, followed by wound migration assay (Fig. 2). While HCT116 (p53−/−) cells transfected with CtBP2 siRNA showed the expected drastic reduction in basal cell migration activity as compared to the control, cells transfected with CtBP2 siRNA followed by Tiam1 overexpression demonstrated a partial reversal of the block in cell migration caused by CtBP2 depletion, with significant restoration of nearly 50% of the normal migratory activity compared with CtBP2 knockdown alone (P < 0.05) (Fig. 2A). Both CtBP2 knockdown and overexpression of the Tiam1 protein were confirmed by immunoblots (Fig. 2A, right panel). This experiment also reinforces the finding that CtBP2 depletion leads to a reduction in Tiam1 levels (Fig. 2A, right panel).
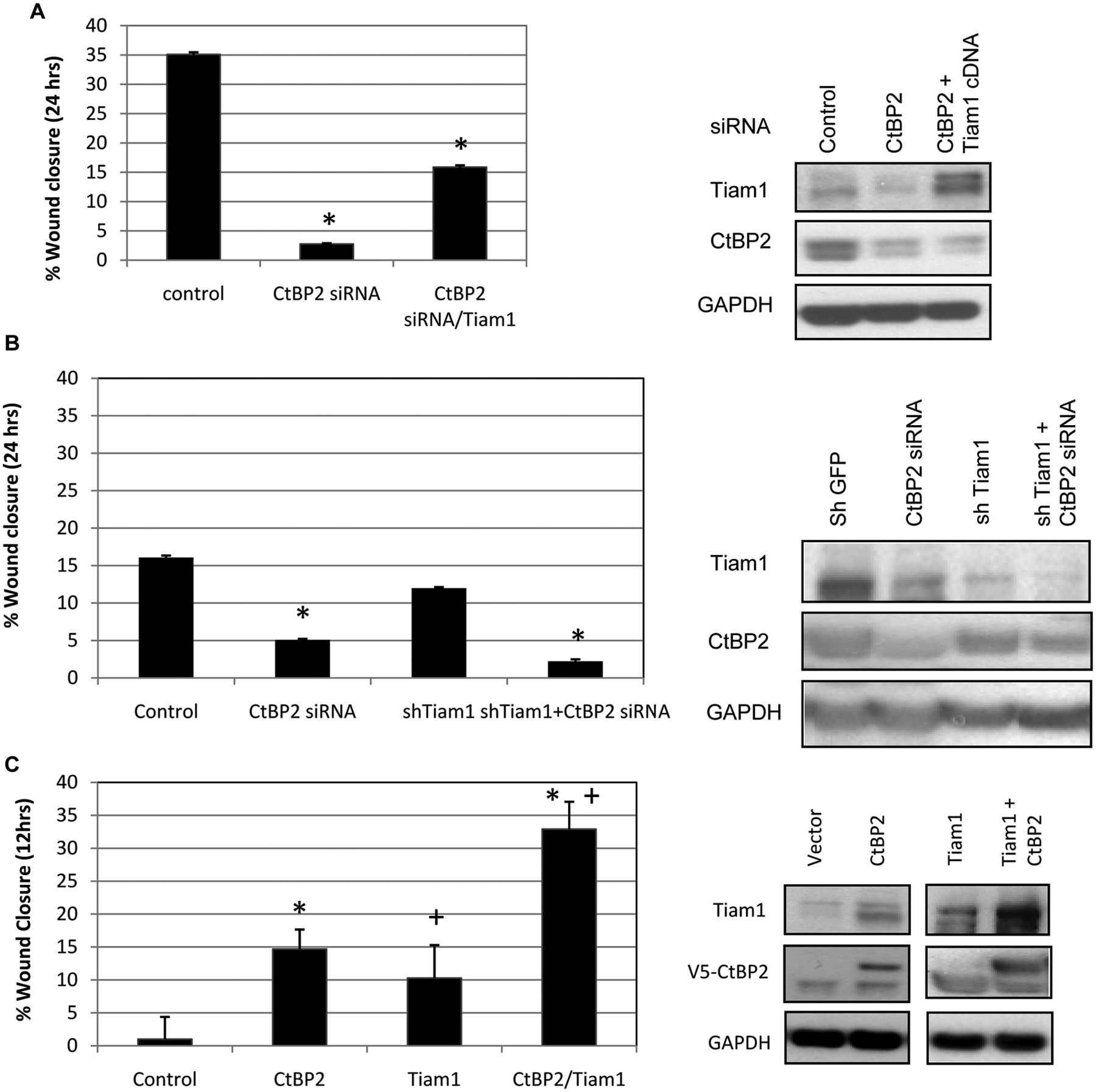
Functional interplay of CtBP2 and Tiam1 in regulating colon cancer cell migration. (
Functional interplay of CtBP2 and Tiam1 in the regulation of cell migration
To determine if CtBP2 and Tiam1 regulate cell migration through the same functional pathway, CtBP2 and Tiam1 were depleted individually or in combination using siRNA and shRNA, respectively, or overexpressed individually or in combination, following which cell migration was analyzed. The effect of CtBP2 and Tiam1 depletion individually resulted in the inhibition of HCT116 (p53−/−) cell migration, with CtBP2 knockdown exerting a more profound inhibition (70% reduction) than Tiam1 knockdown (25% reduction; P < 0.05) when either was compared to control siRNA (Fig. 2B, left panel). Notably, further abrogation of cell migration was observed with the simultaneous knockdown of both CtBP2 and Tiam1 (~90% reduction; P < 0.05 for comparison of shTiam1 + siCtBP2 v. siCtBP2) (Fig. 2B, left panel). Immunoblots confirmed the appropriate individual effect of Tiam1 and CtBP2 siRNA in each of the cell lysates, and the combination of CtBP2 siRNA with Tiam1 shRNA resulted in the additional depletion of Tiam1, consistent with an activator function for CtBP2 in Tiam1 expression (Fig. 2B, right panel). Thus, both CtBP2 and Tiam1 contribute directly to HCT116 (p53−/−) cell migration, although the difference in the biological effect of CtBP2 versus Tiam1 knockdown, which both result in similar Tiam1 levels, indicates that CtBP2 likely exerts its effects on migration through both Tiam1-dependent and -independent pathways. 2
To further investigate the hypothesis that direct regulation of Tiam1 expression by CtBP2 results in migratory phenotypes, CtBP2 or Tiam1 was overexpressed individually, or in combination, in HCT116 (p53−/−) colon cancer cells, followed by migration assay. Cell migration increased 10- to 15-fold in colon cancer cells transfected individually with CtBP2 or Tiam1 expression vectors (Fig. 2C). However, cells co-transfected with CtBP2 and Tiam1 together showed a significant additive (2- to 3-fold) increase in cell migration as compared to the expression of each factor alone (P < 0.05 for comparison of CtBP2 + Tiam1 expression v. either expressed alone), suggesting that CtBP2 and Tiam1 cooperate in regulating cell migration (Fig. 2C). Immunoblotting verified the overexpression of CtBP2 and Tiam1, and interestingly, the levels of Tiam1 achieved were equivalent after Tiam1 or CtBP2 overexpression. With both factors overexpressed, there was a significant additional increase in Tiam1 expression correlating with the additional increase in migration seen when both were overexpressed (Fig. 2C, right panel).
CtBP2 regulation of the Tiam1 promoter through KLF8 recognition elements
Given the possibility that CtBP2 was acting as a transcriptional activator of the Tiam1 gene, the Tiam1 promoter region was analyzed for recognition sites relevant to CtBP binding transcription factors. Three sites with an exact match to KLF8 (ZNF741/BKLF3) recognition elements 29 were identified within the −1 to −2,200 upstream promoter region of the Tiam1 locus (Fig. 3A). We therefore hypothesized that KLF8 recruits CtBP2 to the Tiam1 promoter and that this recruitment in turn regulates Tiam1 promoter activity. To test this hypothesis, a luciferase reporter containing the Tiam1 promoter was co-transfected into HCT116 (p53−/−) cells with CtBP2, or CtBP2 and KLF8 expression vectors, as indicated (Fig. 3B). Co-expression of CtBP2/KLF8 activated the Tiam1 promoter by approximately 1.5-fold (P < 0.05) (Fig. 3B), whereas there was no significant activation of the Tiam1 promoter after the expression of CtBP2 alone. Neither CtBP2 alone nor CtBP2 in combination with KLF8 expression vectors enhanced the expression of a control reporter lacking KLF8 recognition elements (Fig. 3B).
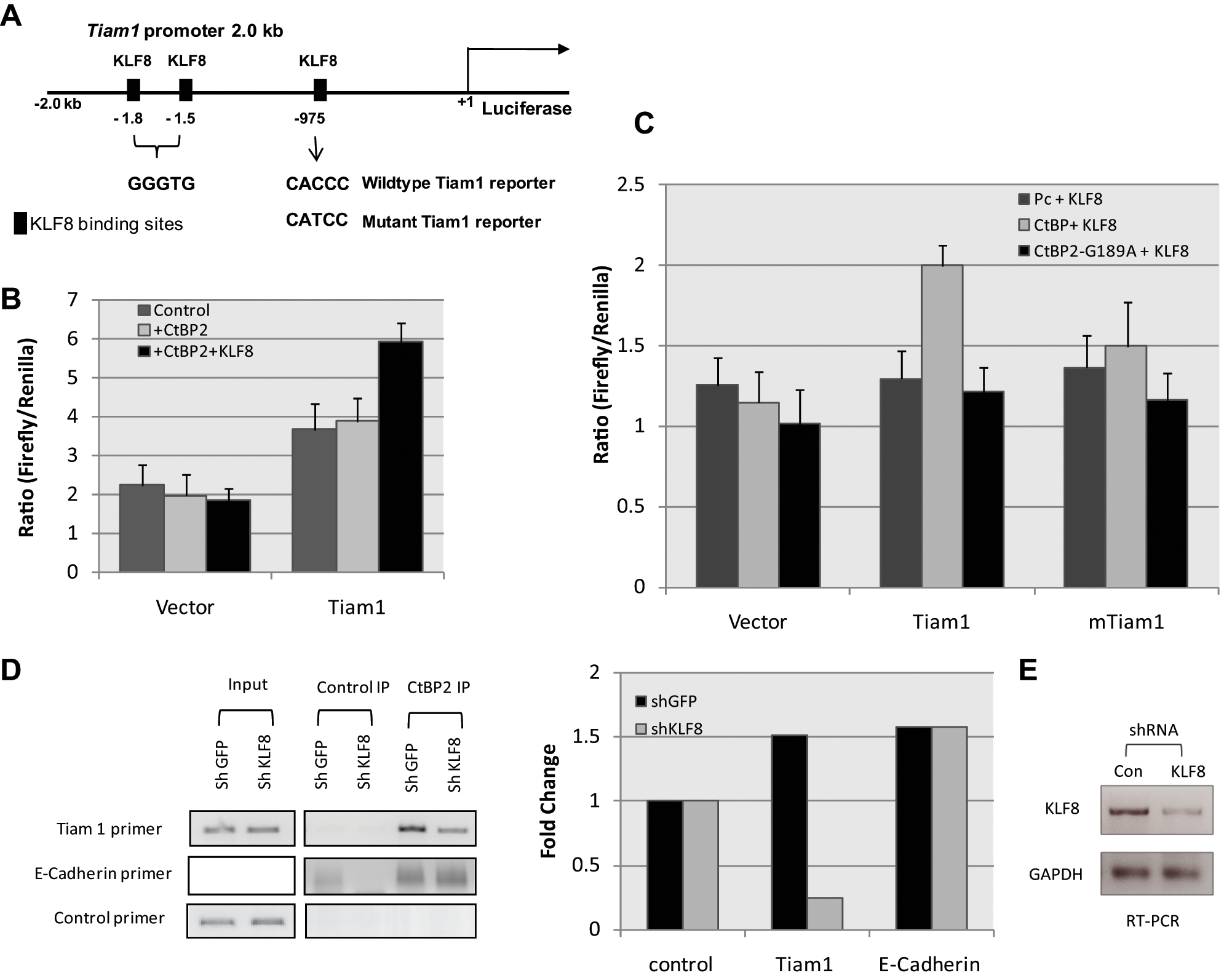
Tiam1 is a transcriptional activation target of CtBP via KLF8 binding. (
To confirm that intact KLF8 sites were necessary for CtBP2 regulation of Tiam1, Tiam1 promoter luciferase reporters containing either all wild-type KLF8 binding sites or with the KLF8 binding site nearest to the transcription start site mutated were transfected into HCT116 (p53−/−) cells along with expression vectors for 1) KLF8, 2) CtBP2 and KLF8, or 3) mutant CtBP2 (defective in NADH binding) and KLF8 (Fig. 3C). Co-expression of CtBP2 with KLF8 resulted in an approximately 1.5-fold increase in wild-type Tiam1 promoter activity (P < 0.05), while the mutant Tiam1 promoter was not activated by combined CtBP2/KLF8 expression (Fig. 3C). This suggests that the KLF8 binding site is required for CtBP2/KLF8 activation of the Tiam1 promoter. Further, when a mutant CtBP2 defective for NADH binding and co-repressor activity 2 was co-transfected with KLF8, the combination had no effect on Tiam1 promoter activity, suggesting that CtBP2 and its NADH binding activity, in concert with KLF8, is required for co-activation of Tiam1 (Fig. 3C). Thus, KLF8 elements and the CtBP2 NADH binding domain are crucial for CtBP/KLF8-mediated regulation of the Tiam1 promoter.
CtBP2 is recruited to the Tiam1 promoter
Previous studies have shown that CtBP2 binds to KLF8 and regulates the expression of genes downstream of KLF8 recognition elements.29,30 To address whether CtBP2 is directly recruited to the Tiam1 promoter, CtBP2 ChIP was performed using chromatin from HCT116 (p53−/−) colon carcinoma cells and either control IgG or anti-CtBP2 antibody (Fig. 3D). The Tiam1 PCR primer set amplified a fragment encompassing the proximal KLF8 binding site at -975bp relative to the transcription start site of the TIMA1 promoter that is critical for CtBP regulation of the promoter (Fig. 3A). A control primer set amplified a fragment located 10 kb upstream of the Tiam1 promoter, whereas the E-cadherin promoter primer set was used as a positive control for a promoter occupied by CtBP2 but not via KLF8 sites 31 (Fig. 3D). To also test the hypothesis that KLF8 was directly targeting CtBP2 to the Tiam1 promoter, the chromatin for these analyses was prepared from HCT116 (p53−/−) cells expressing either a control (shGFP) or KLF8-specific shRNA (sh-KLF8). The efficacy of KLF8 knockdown was determined by RT-PCR (Fig. 3E). Equally robust CtBP2 binding was observed with both the Tiam1 and E-cadherin promoter regions in chromatin from shGFP cells interrogated by the respective primer sets (Fig. 3D). Adding specificity to these data, CtBP2 did not localize to the control fragment, and the control IgG ChIP with the Tiam1 primer set also did not yield any signal (Fig. 3D). Consistent with a CtBP- targeting role for KLF8 at the TIAM promoter, CtBP2 recruitment to the Tiam1 promoter was reduced in the absence of KLF8 (consistent with the level of its knockdown) (Fig. 3E). KLF8 depletion had no effect on CtBP2 recruitment to the E-cadherin promoter, as CtBP is recruited to the E-cadherin promoter by E-box factors (Twist, Snail1,32,33) rather than KLF8 (Fig. 3D).
Tiam1 as a transactivation target and downstream effector of CtBP-induced cell migration
CtBP acts by transcriptionally regulating several genes involved in major pathways promoting cancer, as identified in a wide range of studies in humans, mice, and Drosophila.6,10,34 CtBPs have a direct role in stimulating cell migration and invasion, a characteristic that is linked to its prometastatic function in cancer.2,3 In this report, we demonstrated that CtBP2 transcriptionally activates Tiam1 to promote colon cancer cell migration. Tiam1 was found to be invariably upregulated in cells overexpressing CtBP2 and downregulated with CtBP depletion. This effect was observed both at the protein and mRNA expression levels, and a transcriptional analysis identified Tiam1 as a transcription activation target of CtBP2.
We found CtBP2 to be recruited to the Tiam1 promoter largely through KLF8, as is the case for CtBP recruitment to the Bik promoter. 31 This suggests that Tiam1 is a transcriptional activation target of CtBP via KLF8 binding and that the KLF8 elements are crucial for CtBP/KLF8-mediated activation of the Tiam1 promoter. Tiam1 is therefore a novel target for CtBP2 transcriptional regulation, but unlike its previously well-understood co-repression role, CtBP2 here is playing a paradoxical but previously described role as a transcription activator.34-38 Notably, in H1299 cells but not HCT116 cells, the changes in the Tiam1 protein were consistently greater (positive or negative) than the changes in mRNA expression, suggesting that in these cells, additional posttranscriptional mechanisms may also be at play in the CtBP regulation of Tiam1 protein abundance.
CtBP2 transcriptional activation of Tiam1 resulted in increased colon cancer cell migration, as HCT116 (p53−/−) colon cancer cells transfected with CtBP2 siRNA followed by Tiam1 overexpression demonstrated rescue of the block in cell migration caused by CtBP2 depletion. Thus, CtBP2 depletion reduced cell migration by reducing Tiam1 levels, and overexpression of Tiam1 in these cells rescued migration. Furthermore, HCT116 (p53−/−) colon carcinoma cells underwent increased migration in response to CtBP2, and exogenous Tiam1 markedly accelerated this process.
CtBP engages multiple pathways to induce cell migration
We have previously reported that CtBP2 potently promotes cell migration by decreasing PTEN levels. Lower PTEN abundance correlates with activation of the PI3K pathway, a major regulator of cell migration via generation of PIP3, which can activate the GEF P-Rex as well as modulate other GEFs and GTPase-activating proteins (GAPs) to activate Rho family GTPases.2,39 In this report, Tiam1 activation by CtBP2 was identified to also play a critical and indispensable role in CtBP2-induced cell migration, although the impact of CtBP2 depletion was more potent on migration than that of Tiam1 depletion despite equal levels of Tiam1 depletion in both cases (Fig. 2B). This would be consistent with CtBP regulating migration by multiple pathways, as it can activate the promigratory PI3K pathway (via PTEN repression) simultaneously with upregulating Tiam1 (Fig. 4). This model raises the interesting possibility that migration is quantitatively regulated by the summed activity of multiple GEFs and GAPs that act on Rho family GTPases. Additionally, CtBP2 depletion’s more potent impact on migration than Tiam1 depletion could also be due, in part, to the CtBP2 transcriptional activation of other Tiam1-related proteins, most importantly its paralog Tiam2, which encodes a similar GEF activity.40,41
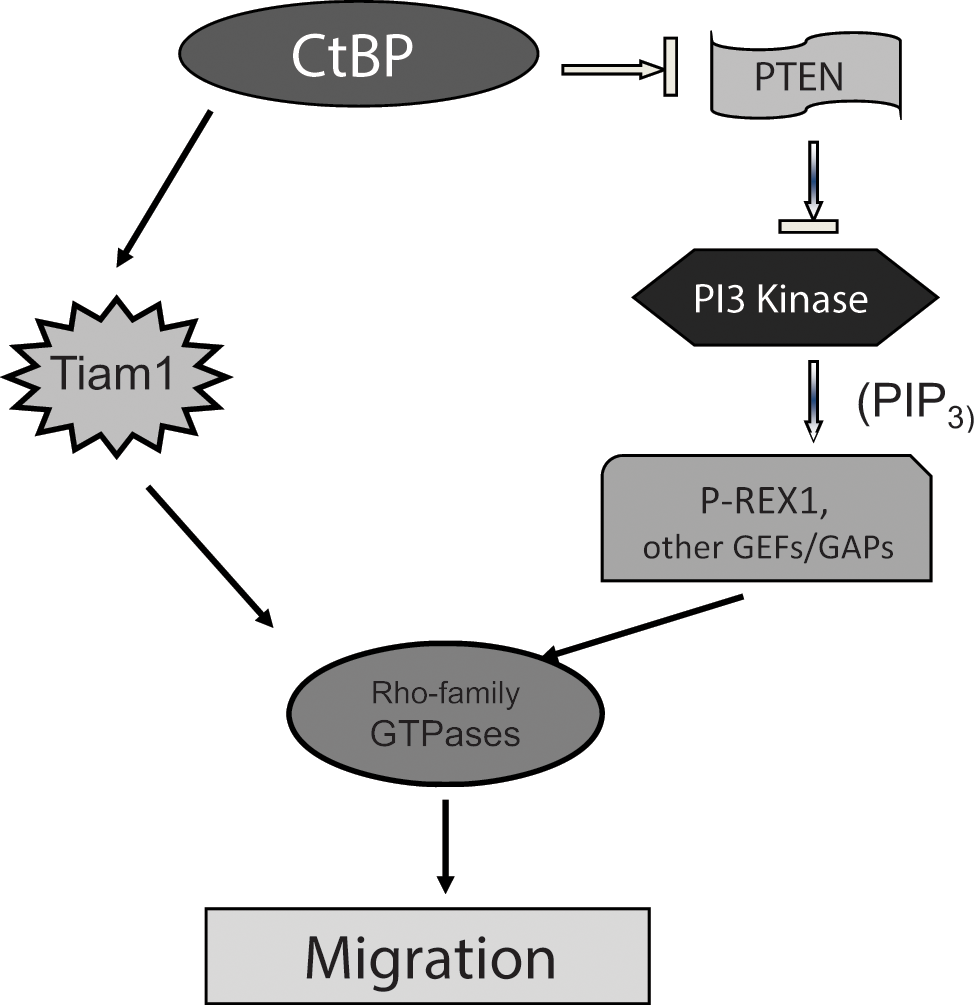
Proposed model for the role of CtBP in cell migration. CtBP coordinately regulates cancer cell migration via repression of PTEN, activating the PI3K pathway,2,39 and also by direct transcriptional activation of the GEF Tiam1 (this work). Both PI3K and Tiam positively regulate Rac and other Rho family small GTPases via GEF activity (intrinsic to Tiam; induced by PI3K via PIP3 regulation of P-Rex and other GEFs) and/or modulation of GAP activities.
Role of Tiam1 in migration/invasion and metastasis
Tiam1 associates with activated, oncogenic Ras-GTP to stimulate Rac activity, and CtBP potently induces activated Rac and migratory/invasive behavior in cancer cell lines. 2 Tiam1 has been directly implicated in cell adhesion and migration, and its activation is associated with increased tumor invasiveness. 28 Specifically, Tiam1 overexpression contributes to colon carcinoma growth at metastatic sites in an orthotopic nude mouse model, 27 while Tiam1 knockdown in a colorectal cancer cell line resulted in the effective inhibition of in vitro growth and invasive ability of colorectal cancer cells and inhibition of in vivo growth of lung and liver metastases. 42 Thus, the promigration function of CtBP2 could contribute to colon cancer metastasis through transcriptional regulation of Tiam1, and Tiam1 may mediate CtBP’s proinvasive and prometastatic functions.3,11
CtBP as a transcriptional activator
CtBP family proteins have been well characterized for their role as transcriptional co-repressors for many transcription factors. 34 This study expands the transcriptional role of CtBP2 as an activator in addition to its well-defined role as a transcriptional co-repressor. Previous reports have suggested that in addition to its canonical repressor activity, CtBP can also mediate transcription activation in certain contexts. 34 In mice, the expression of the Wnt3A target gene Brachyury was abnormally low in E10.5 CtBP2−/− embryos compared to E9.5 embryos, 43 suggesting that mCtBP2 could function as a direct or indirect transcriptional activator of Brachyury. In Drosophila, dCtBP co-activates the transcription of certain Wnt target genes after stimulation with Wnt, even while repressing other Wnt target genes in the absence of Wnt. 36 hCtBP1 was also recently identified as an activator of MDR1 gene transcription in human multidrug-resistant cancer cells. 38 Our results support a model where CtBP is a gene-specific regulator of cell migration, with some targets such as PTEN and Bik undergoing CtBP repression, 2 while other targets such as Tiam utilize CtBP for activation of their expression (Fig. 4).
As CtBP-based therapeutics are developed, 44 and with increasing evidence for aberrant Tiam activity in cancer, Tiam as a GEF enzyme appears to be an attractive target for anticancer drug discovery. The simultaneous inhibition of both Tiam and CtBP may lead to potent effects, especially in the realm of inhibiting cancer cell invasion and metastasis.
Materials and Methods
Cell cultures and transfections
DMEM and McCoy’s medium (Invitrogen, Carlsbad, CA) were supplemented with 10% fetal bovine serum and 100 U/mL of penicillin. Human H1299 (lung carcinoma; p53 null) cells were cultured in DMEM, and HCT116 human colon cancer cells (ARF silenced) with a targeted deletion of p53 (p53−/−) 45 were grown in McCoy’s medium (Invitrogen). Cells were incubated at 37°C in a humidified, 5% CO2 environment. Expression plasmids were transfected using Fugene (Roche, Basel, Switzerland), and small interfering RNA (siRNA) duplexes were transfected using Oligofectamine (Invitrogen). The siRNA concentration used for transfection was 40 nmol/L, except where noted. The siRNA sequence for human CtBP2 (hCtBP2) was 5′-AAGCGCCUUGGUCAGUAAUAG-3′ and for shKLF8 was 5′-CTGGTCGATATGGATAAACTCA-3′. A human Tiam1 shRNA clone was obtained from Open Biosystems (Huntsville, AL). Nontargeting control siRNA (IX) was obtained from Dharmacon (Lafayette, CO). Vector alone, Tiam1, CtBP2, or CtBP2 mutant–expressing stable H1299 cells were generated using geneticin selection. shGFP and shTiam1-expressing stable H1299 cells were generated using puromycin selection.
Plasmids
Pc-V5CtBP2, Pc-V5CtBP2-G189A, and Pc-V5BKLF have been described.2,12,31 Pc-Tiam1 expression plasmid was the generous gift of A. Mercurio.
Antibodies and Western blotting
Cells were lysed in lysis buffer (20 mM HEPES, pH 7.4, 0.5% Triton X-100, 2 mM MgCl2, 10 µM ZnCl2, 2 mM N-ethylmaleimide, 1 mM phenylmethylsulfonyl fluoride, and 240 mM NaCl) containing complete Mini Protease Inhibitor Cocktail Tablets (Roche). Antibodies used were as follows: CtBP2 (BD Transduction Laboratories, Franklin Lakes, NJ), hARF (Abcam, Cambridge, UK), V5 tag (Invitrogen), Tiam1 (Santa Cruz Biotechnology, Santa Cruz, CA), and GAPDH (Advanced ImmunoChemical, Long Beach, CA). Anti-rabbit IgG–horseradish peroxidase and anti-mouse IgG–horseradish peroxidase conjugates (Amersham, Amersham, UK) were used with enhanced chemiluminescence detection (Amersham) for Western blotting.
qRT-PCR
mRNA transcripts for human Tiam1 and GAPDH were analyzed by qRT-PCR using SYBR green (Applied Biosystems, Foster City, CA) and an ABI 7300 (Applied Biosystems). Relative amounts of the mRNA transcripts were calculated using the ΔΔCT method with GAPDH and β-actin mRNA as internal references. qRT-PCR for Tiam1 and GAPDH was performed using the following primers:
Tiam1
sense: 5′-AAGACGTACTCAGGCCATGTCC-3′
antisense: 5′-GACCCAAATGTCGCAGTCAG-3′
GAPDH
sense: 5′-ATCACCATCTTCCAGGAGCGA-3′
antisense: 5′-GCCAGTGAGCTTCCCGTTCA-3′.
Cell motility and migration assays
For wound assays, equal numbers of cells were plated at 50% confluency in 6-well plates, and 24 hours after plating, the cells were transfected with either siRNA or expression plasmids. Twenty-four hours after transfection, the confluent monolayer of cells was scraped to introduce a wound, and the cells were incubated for another 12 or 24 hours. Captured images were used to compare and quantify the percentage of wound closure by measuring the migration distance of cells found in the cleared area from the point the cells were scraped to the migration front. Five independent 100× fields were measured for each experiment as described, 2 and the values were normalized to the negative control set, which was set at 100% for each experiment. The percentage of wound closure and standard deviation were calculated for 3 experiments.
Tiam promoter luciferase reporter assay
A 2.0-kb region of the human Tiam1 promoter (−2,200 to −267) was amplified by PCR from human genomic DNA and inserted into the firefly luciferase reporter, pGL3 (Promega, Madison, WI). A KLF8 binding defective mutant Tiam1 promoter plasmid was generated from the pGL3-Tiam1 wild-type luciferase reporter plasmid using the QuikChange PCR protocol (Stratagene, La Jolla, CA). HCT116 cells were transfected with 1) pGL3-Tiam1, 2) pGL3-mTiam1, and 3) a control plasmid expressing the Renilla luciferase (pRL-TK), with vector alone, CtBP2, or an NADH binding defective allele (G189A) of CtBP2 together with KLF8. The expression of reporter genes was determined using a Dual Luciferase assay kit (Promega) after 24 hours of transfection.
ChIP assay
Cells were plated for 24 hours, and approximately 108 cells were used for each ChIP assay. Cells were washed once in PBS and were treated with 1% formaldehyde in cold PBS for 10 minutes at 4°C with continuous shaking. Glycine (final concentration of 125 mM) was added to quench the formaldehyde for 5 minutes at 4°C with continuous shaking. Cells were then harvested and washed twice with ice-cold PBS. Nuclei were isolated by incubating the cells in nucleus isolation buffer (5 mM PIPES, pH 8.0, 85 mM KCl, and 0.5% NP-40) for 20 to 30 minutes on ice. The nuclei were harvested at 4°C by centrifuging the cell suspension at 7,000g for 5 minutes and then resuspending the pellet in 2 mL of RIPA buffer (150 mM NaCl, 1% v/v NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0, and 5 mM EDTA) containing complete Mini Protease Inhibitor Cocktail Tablets (Roche). Chromatin was fragmented to approximately 200 to 700 bp by sonication. Nuclear debris was removed by centrifuging the lysates at 4°C for 15 minutes at 14,000 rpm. The lysate was precleared by incubation with protein G Sepharose beads (Sigma-Aldrich, St. Louis, MO) for 30 minutes at 4°C. Immunoprecipitation was then performed overnight at 4°C with control (mouse IgG) and CtBP2 antibody, protein G Sepharose beads (Sigma-Aldrich) were added, and the immunocomplexes were then allowed to bind to the beads for 2 hours at 4°C. The beads were washed once each with RIPA buffer, RIPA buffer with 500 mM NaCl, immunoprecipitation wash buffer (10 mM Tris-HCl, pH 8.0, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, and 1 mM EDTA), and finally with Tris-EDTA. Beads were resuspended in 200 µL of elution buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, and 1% SDS) with Proteinase K and incubated overnight at 55°C. DNA was extracted using phenol chloroform, precipitated in the presence of glycogen by ethanol, allowed to air dry, and dissolved in 10 mM Tris-Hcl (pH 8.0), 1 mM EDTA. The immunoprecipitated DNA was diluted 10-fold to ensure that the amplified product was in the linear range following amplification by PCR. The following sets of primers were used to amplify different regions of the indicated genes:
Tiam1, primer set 1
sense: 5′-AATTAGGCAGCCTCACTCCA-3′
antisense: 5′-CCCCTGAAATCTGTTCTCCA-3′
Tiam1, primer set 2
sense: 5′-CGTGTTCTGGGGAAAGAAAA-3′
antisense: 5′-AGGACTCCCAGGGTTCACTT-3′
nonspecific irrelevant primers 20 kb upstream of KLF8 binding sites
sense: 5′-ACCTTGCCTGGCCTTATTTT-3′
antisense: 5′-CCTGAGGTCAGGAGTTCGAG-3′
E-cadherin
sense: 5′-TAGCCTGGCGTGGTGGTGTG CACCTG-3′
antisense: 5′-GTGCGTGGCTGCAGCCAGGT GAGCC-3′.
RT-PCR analysis was done to confirm KLF8 knockdown from HCT116 cells expressing either a control (shGFP) or KLF8-specific shRNA (sh-KLF8). RT-PCR was performed using the following primers:
KLF8
sense: 5′-AGGTGGCTCAATGCAGGTAT-3′
antisense: 5′-CATGGGCAGAGACTGCACTA-3′
GAPDH
sense: 5′-ATCACCATCTTCCAGGAGCGA-3′
antisense: 5′-GCCAGTGAGCTTCCCGTTCA-3′.
Footnotes
Acknowledgements
The authors thank A. Mercurio and members of the Mercurio laboratory for assistance with reagents and for helpful discussions. The authors also thank Dr. Kevin Hogan for his comments and editorial assistance with the article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: S.R.G. was supported by an ACS Research Scholar Award, and S.P. was supported by the Pancreatic Cancer Alliance.