
Abstract
It has become increasingly clear that microRNAs (miRNAs) play important roles in tumorigenesis and metastasis. Recently, miR-203 was reported as a suppressor microRNA often silenced in different malignancies including hepatocellular carcinoma, prostate cancer, oral cancer, and hematopoietic malignancy, but little is known about its potential role in breast carcinogenesis. In this study, we found that in breast cancer, miR-203 was upregulated in primary tumors and some nonmetastatic cell lines but was significantly downregulated in metastatic cell lines including BT549, Hs578T, and MDA-MB-231, as measured by regular and real-time PCR. Downregulation of miR-203 in metastatic breast cancer cells appeared to be caused by hypermethylation of its promoter. Functionally, ectopic expression of miR-203 in BT549 and MDA-MB-231 breast cancer cell lines caused cell cycle arrest and apoptosis and inhibited cell invasion and migration in vitro. Bioinformatic analysis predicted the snail homolog 2 (SNAI2 or SLUG), a transcription factor that promotes cell invasion and tumor metastasis, as a target of miR-203, and the prediction was validated by expression analysis and luciferase reporter assay of the 3′ untranslated region of SNAI2 that contains the miR-203 target sequences. These results suggest that in malignant breast cancer cells, miR-203 is epigenetically silenced, and the silencing promotes tumor cell growth and invasion at least in part by upregulating the SNAI2 transcription factor.
Introduction
MicroRNAs (miRNAs) are a class of endogenous small RNA molecules that are highly conserved in a variety of eukaryotic organisms. Recent studies have shown that miRNAs play important roles in multiple cellular processes including proliferation, apoptosis, differentiation, senescence, organ development, and tumorigenesis. About 50% of human miRNAs are frequently located at or near fragile sites or cancer-associated genomic regions. 1 miRNAs involved in tumorigenesis can act as either oncogenes or tumor suppressor genes. miR-203 is a stemness-inhibiting miRNA that induces epidermal differentiation by restricting proliferative potential and targeting the stemness-related transcription factor ΔNp63.2,3 Its abnormal expression has been detected in several types of human cancers, including frequent upregulation in bladder and ovarian cancers4,5 and association of overexpression with poorer patient survival in colon and pancreatic cancers.6,7 In the squamous type of carcinomas and hepatocellular carcinoma, however, miRNA-203 expression is reduced, and DNA hypermethylation appears to be responsible for the downregulation.8-11 Downregulation of miRNA-203 has also been detected in animal models of oral squamous cell carcinoma and T cell lymphoma.12,13 In prostate cancer cell lines DU 145 and PC-3, overexpression of miR-203 is sufficient to induce a mesenchymal-to-epithelial transition with the inhibition of cell proliferation, migration, and invasion. 14 At the molecular level, the transcriptional repressor zinc-finger E-box binding homeobox 1 (ZEB1) has been identified as a key inducer of EMT that promotes invasion and metastasis in different types of human tumors. 15 One mechanism by which ZEB1 activates EMT appears to be the downregulation of several miRNAs including miR-203.16-18 These results suggest that miR-203 plays a role in tumorigenesis, but whether it modulates the initiation stage or the progression stage remains to be clarified.
Breast cancer is one of the most common malignances and leading causes of death for women all over the world. Lethal invasion is a major characteristic of metastatic cancer cells, but the mechanisms of tumor invasion and metastasis are still not well understood. SNAI2 (also named Snail2 or SLUG), a member of the Snail family of C2H2-type zinc-finger transcription factors, promotes epithelial-mesenchymal transition and shows antiapoptotic activity. 19 In breast cancer, SNAI2 is induced by Twist1, and the induction is essential for Twist1 to induce cell invasion and tumor metastasis, 19 which involves the repression of several factors in breast cancer cells.20,21
In this study, we determined whether miR-203 plays a role in human breast cancer and found that the expression of miR-203 was increased in primary tumors and nonmetastatic cell lines but was downregulated in metastatic breast cancer cell lines. Promoter methylation appeared to be responsible for the downregulation of miR-203. Functionally, re-expression of miR-203 in metastatic breast cancer cell lines not only suppressed cellular invasion and motility but also caused cell cycle arrest and apoptosis. Mechanistically, the EMT-promoting transcription factor SNAI2 was identified as a target of miR-203. These results suggest that miR-203 could play an inhibitory role in metastatic progression of breast cancer.
Results
Expression of miR-203 is upregulated in breast cancer primary tumors and nonmetastatic cell lines but significantly downregulated in metastatic cell lines. We first evaluated the expression of miR-203 by real-time PCR in a group of primary breast cancer tumors, with the matched noncancerous tissue from the same patient as the control. Among the 36 cases examined, 18 (50%) showed an increase in miR-203 expression by at least 1-fold, as their tumor/normal ratios of miR-203 expression were at least 2 (mean, 9.3), whereas 13 (36%) showed a tumor/normal ratio between 1 and 2 (mean, 1.4), and only 5 (14%) had a tumor/normal ratio equal to or smaller than 1 (mean, 0.5) (Fig. 1A and Suppl. Table S1). These results suggest that miR-203 is often upregulated in primary breast cancer tumors.
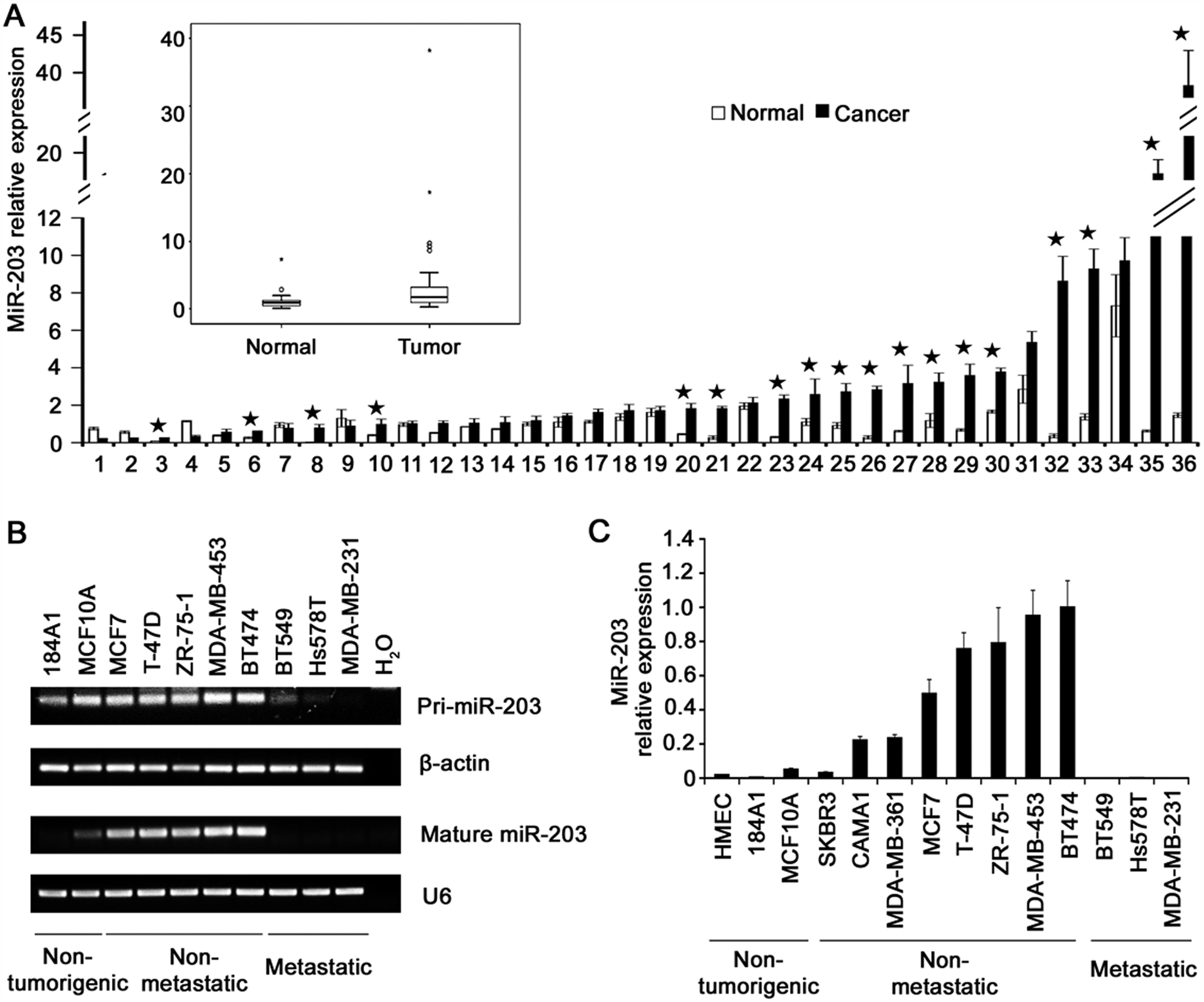
Upregulation of miR-203 in primary tumors and nonmetastatic cell lines and downregulation in metastatic cell lines of breast cancer. (
We then examined the expression of miR-203 in immortalized and nontumorigenic breast epithelial cell lines and breast cancer cell lines with known metastatic potential. Both the primary transcript and mature miR-203 were significantly upregulated in nonmetastatic breast cancer cell lines when compared to immortalized cell lines but were significantly downregulated in metastatic cell lines of breast cancer when compared to immortalized noncancer lines or nonmetastatic cancer lines (Fig. 1B and 1C and Suppl. Table S2).
miR-203 is epigenetically silenced in metastatic breast cancer cell lines. Based on previous studies that demonstrated promoter methylation as the major mechanism for the downregulation of miR-203 in some cancers, we determined whether promoter methylation also caused the downregulation of miR-203 in metastatic breast cancer cell lines. miR-203 has a typical CpG island in its promoter region, which is similar to many tumor suppressor genes (Fig. 2A). PCR and sequencing of sodium bisulfate–treated genomic DNA showed that the promoter of miR-203 was not methylated in the 3 nonmetastatic breast cancer cell lines examined (MCF-7, T-47D, and ZR-75-1) but was moderately methylated in the 3 metastatic cell lines examined (BT549, Hs578T, and MDA-MB-231) (Fig. 2B). In addition, treatment of cells with the 5-aza-dCyd demethylating reagent in both MDA-MB-231 and BT549 cell lines significantly restored the expression of miR-203 (Fig. 2C). For Figure 2C, each data point represents 2 separate experiments conducted in triplicate (MDA-MB-231: P = 1.7 × 10−6; BT549: P = 0.036). These results suggest that downregulation of miR-203 in metastatic breast cancer cells is caused at least in part by promoter methylation.
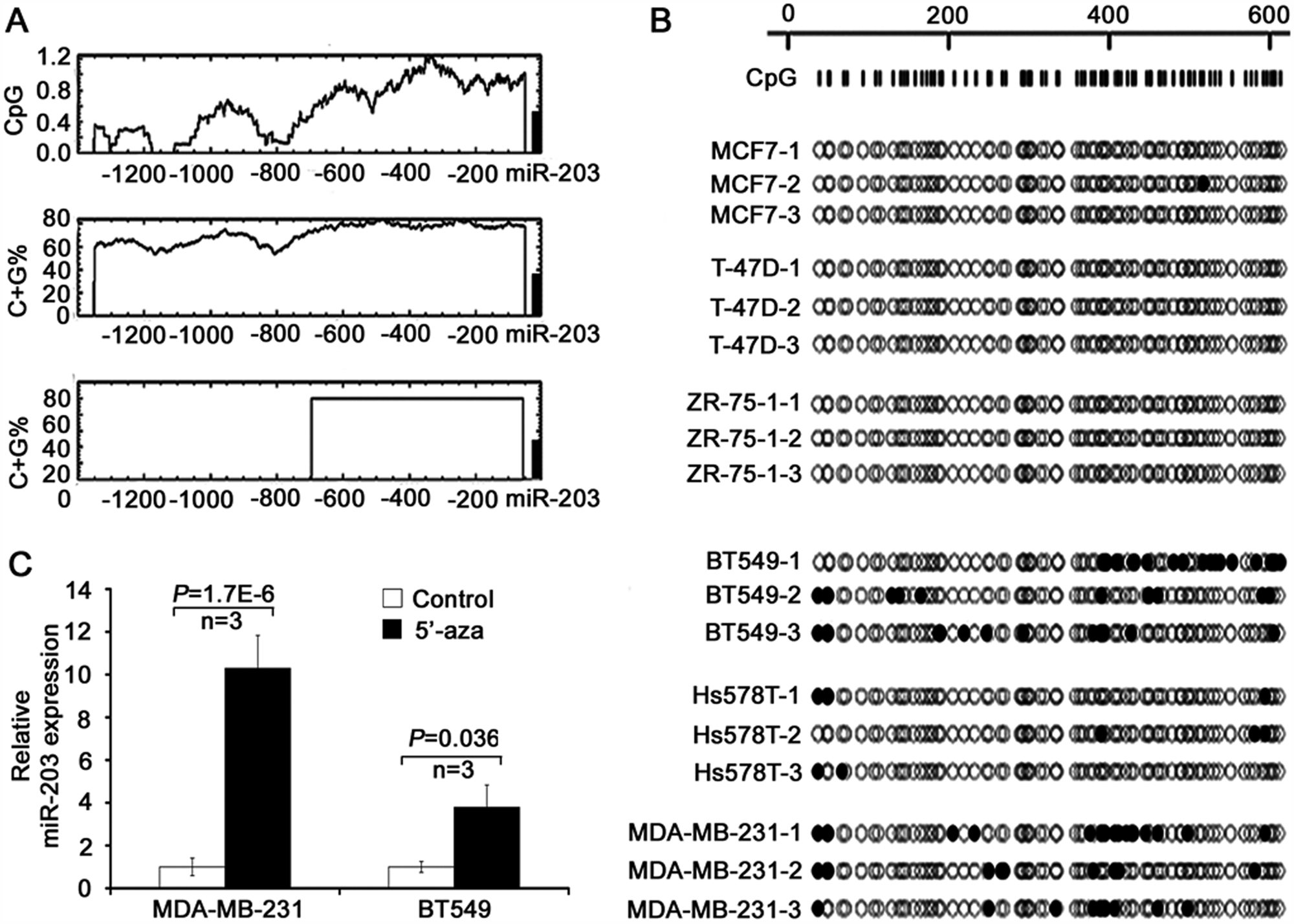
Promoter methylation in the downregulation of miR-203 in metastatic breast cancer cell lines. (
Expression of miR-203 inhibits cell cycle progression and induces apoptosis. To determine whether miR-203 modulates cell proliferation and cell death, we performed in vitro colony formation assay in BT549 and MDA-MB-231 cells transfected with synthetic miR-203 mimics or control mimics. The efficiency of miRNA mimics transfection was confirmed by RT-PCR (Fig. 3A and 3B). Compared to control mimics, expression of miR-203 mimics significantly reduced cell numbers in both BT549 and MDA-MB-231 cell lines (Fig. 3C and 3D), especially in BT549 cells.
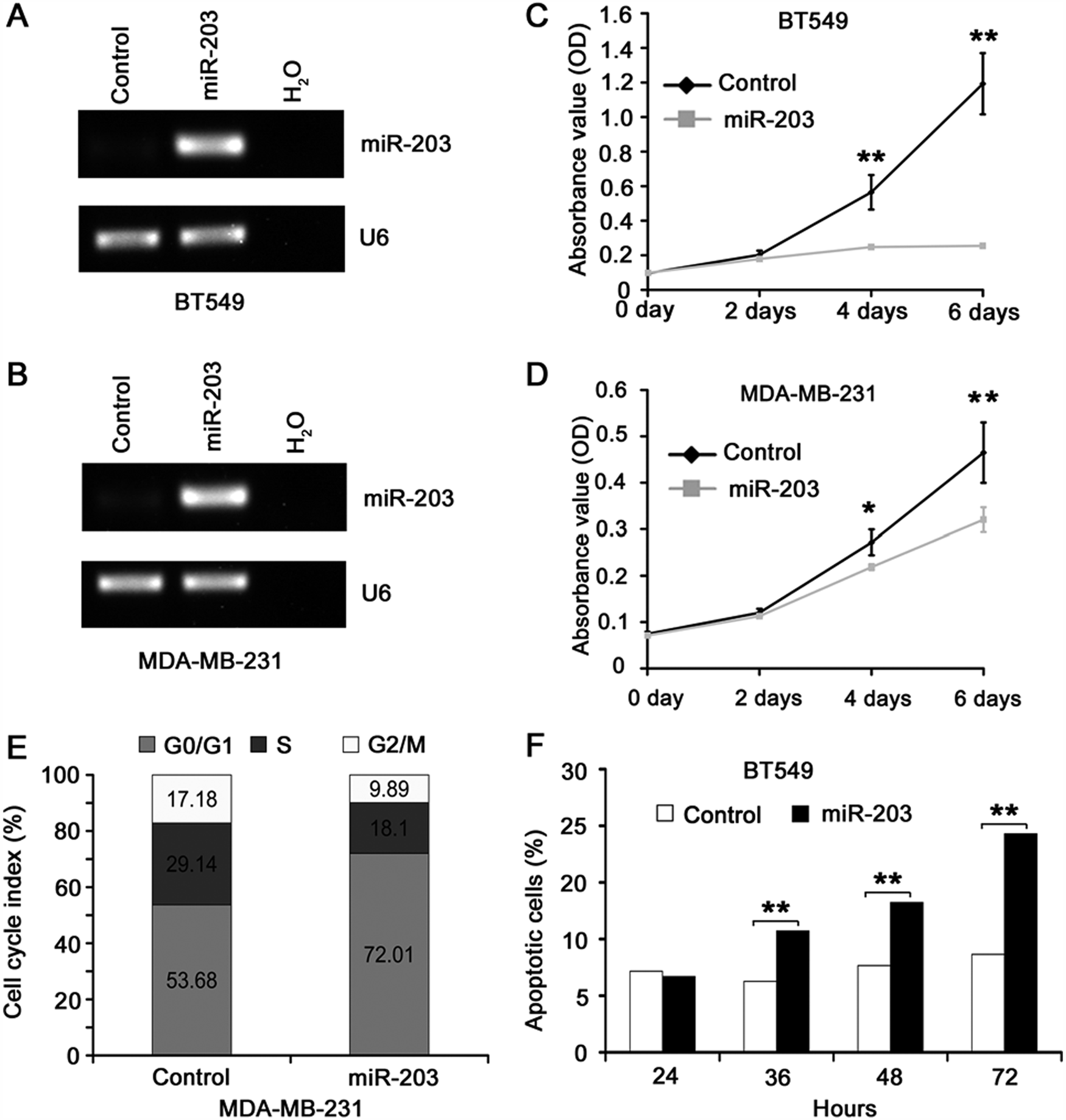
Ectopic expression of miR-203 causes cell cycle arrest in MDA-MB-231 cells and apoptosis in BT549 cells. (
To determine whether the reduction in cell numbers by miR-203 was mediated by cell cycle arrest or apoptosis induction, we first examined the cell cycle distribution of cells by using propidium iodide (PI) staining and flow cytometer approach in both the BT549 and MDA-MB-231 cell lines. The percentage of cells in different phases was compared between miR-203–transfected cells and control cells. Interestingly, whereas expression of miR-203 did not cause noticeable differences in cell cycle distribution in BT549 cells (Suppl. Fig. S1B), it caused significant cell cycle arrest in the G0/G1 phase in the MDA-MB-231 cells (Fig. 3E and Suppl. Fig. S1A). We then examined whether miR-203 causes apoptosis in the 2 cell lines. Whereas expression of miR-203 did not cause obvious apoptosis in MDA-MB-231 cells (Suppl. Fig. S2B), it induced significant apoptotic cell death in BT549 cells in a time-dependent manner (Fig. 3F and Suppl. Fig. S2A). These findings suggest that expression of miR-203 in metastatic breast cancer cells could cause either cell cycle arrest or apoptosis depending on the cell line.
miR-203 suppresses cellular invasion and migration in vitro. To determine whether miR-203 modulates cell migration and invasion directly, BT549 and MDA-MB-231 cells were transduced with miR-203 and control mimics, and wound healing assay was performed. Expression of miR-203 significantly slowed the gap closing in both BT549 (Fig. 4A) and MDA-MB-231 (Fig. 4B) cells. Transwell assay further demonstrated that miR-203 expression suppressed the invasion of both BT549 (Fig. 4C and 4E) and MDA-MB-231 (Fig. 4D and 4F) cells. As shown by growth curves of MDA-MB-231 and BT549 cells transfected with miR-203 and control mimics, cell number did not show obvious changes within 2 days of transfection. Therefore, we limited the time for wound healing and transwell experiments within 48 hours after transfection and/or cell seeding.
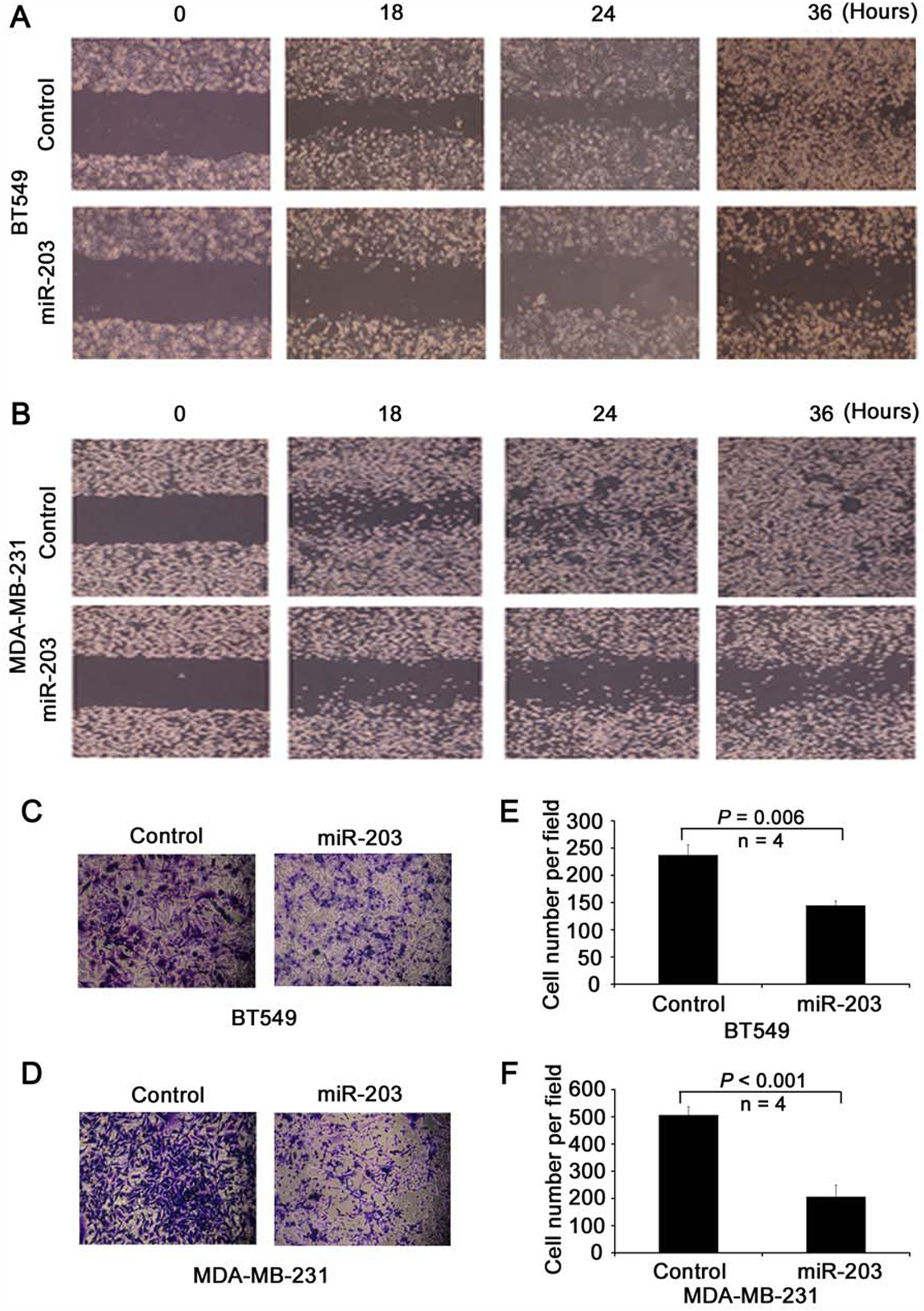
Suppression of cell motility and invasion by miR-203 in metastatic breast cancer cells. (
SNAI2 is a direct target of miR-203. To further explore the mechanism for miR-203–mediated suppression of cell invasion and migration, we searched for targets of miR-203 using some of the well-known miRNA target prediction programs including TargetScan5.1, 22 Pictar, 23 and miRBase Targets. 24 We identified a total of 12 putative miR-203 target genes that may play a role in the regulation of cancer cell invasion and metastasis (Suppl. Table S3), including previously identified BMI1 and RUNX2 genes. Among these genes, the snail homolog 2 (SNAI2 or SLUG) was of particular interest because it showed the highest total context score (Suppl. Table S3), and its role in EMT has been well established in breast cancer. We analyzed its expression in breast cancer cell lines and found an inverse correlation between SNAI2 and miR-203 expressions at the mRNA level by both regular RT-PCR (Fig. 5B) and real-time PCR (Pearson correlation coefficient = −0.82). We then assessed complementarities of miR-203 to the 3′-UTR sequences of SNAI2 and found 2 seed matches of bases 2 to 8 (Fig. 5A). To determine whether miR-203 inhibits SNAI2 expression, miR-203 was transiently transfected into BT549, Hs578T, and MDA-MB-231 cells, and the expression of endogenous SNAI2 was measured by real-time PCR. Seventy-two hours after transfection, miR-203 expression significantly reduced SNAI2 mRNA levels in each of the cell lines examined (Fig. 5C). Furthermore, we examined 2 target genes of SNAI2, E-cadherin and vimentin, in MDA-MB-231 cells transfected with miR-203 and control mimics and found that E-cadherin was upregulated while vimentin was downregulated by miR-203 (Suppl. Fig. S3). We also transfected the pcDNA-pri-miR-203 plasmid into MDA-MB-231 cells to express miR-203 and found significantly reduced expression of SNAI2 at both mRNA and protein levels (Fig. 5D).
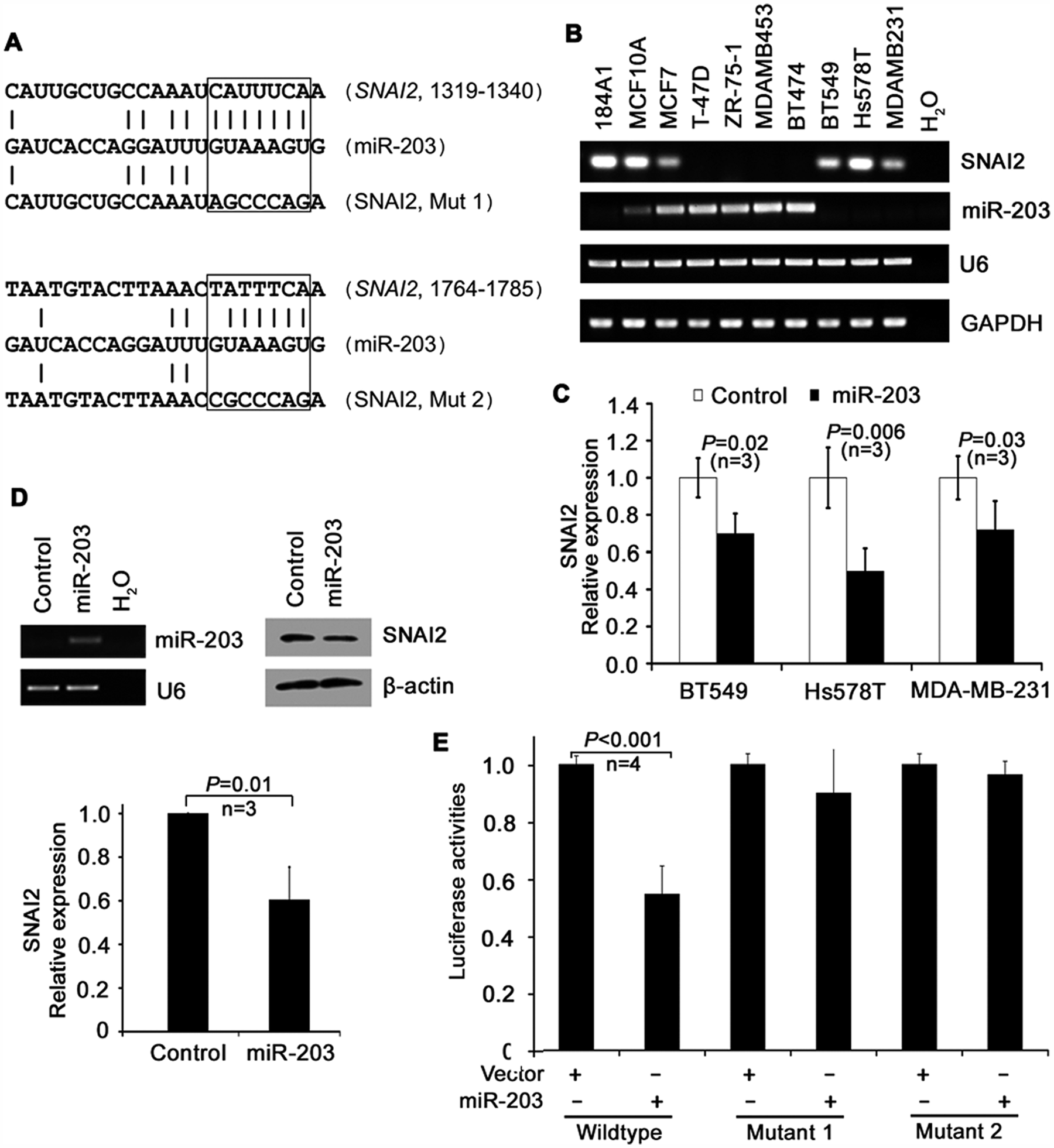
Identification of SNAI2 as a target of miR-203. (
To assess whether miR-203 downregulates SNAI2 through the potential miR-203 target sequences in the 3′-UTR of SNAI2, we cloned the sequence into a luciferase reporter plasmid, made different mutants, and measured the luciferase activities of different constructs in 293T cells. While expression of miR-203 significantly reduced the luciferase activity of the reporter with wild-type sequence, mutation of each of the 2 potential miR-203 target sequences abolished the effect of miR-203 on luciferase activities (Fig. 5A and 5E), indicating that both miR-203 target sequences in the 3′-UTR of SNAI2 are involved in miR-203–mediated regulation.
Discussion
In this study, we examined miR-203 in breast cancer cells and present multiple lines of evidence for a suppressive role of miR-203 in the pathogenesis of breast cancer, including its upregulation in primary tumors and nonmetastatic cell lines but significant downregulation in metastatic cell lines, the induction of apoptosis or cell cycle arrest, the inhibition of migration and invasion, and the downregulation of the EMT-promoting molecule SNAI2. These findings are consistent with findings from other types of cancers, in which miR-203 has been shown to suppress cell proliferation, invasion, and bone metastasis in prostate cancer14,25; to promote apoptosis and inhibit cell proliferation in bladder cancer 26 ; and to inhibit cell proliferation in esophageal cancer. 27
We noticed a difference in the expression of miR-203 in primary tumors between breast cancer and most other types of malignancies examined. In our study, miR-203 was upregulated in the majority (20/36 or 56%) of primary breast cancers when compared to matched noncancerous counterparts from the same patients, and only 3 of the 36 (8%) tumors showed a downregulation in primary cancers (Fig. 1). In published studies of other types of tumors, miR-203 was upregulated in ovarian cancer 4 and downregulated in prostate cancer14,25 and colorectal cancer 28 when compared to noncancerous tissues. Even in the same type of cancer, for example, bladder cancer, one study reported upregulation, 5 and another reported downregulation of miR-203. 26 In pancreatic cancer, miR-203 is mostly upregulated, yet the upregulation appears to be associated with worse patient survival. 29 Taken together with our finding that miR-203 was dramatically upregulated in nonmetastatic breast cancer cell lines when compared to nontumorigenic cells but was absent in metastatic cell lines (Fig. 1), we speculate that miR-203 is upregulated in primary breast cancers to restrain metastatic behavior, and downregulation of miR-203 promotes metastasis.
In this study, we also analyzed the relationship between miR-203 expression and clinicopathological features of breast cancer in the 36 tumor samples. Expression change of miR-203 was not significantly associated with any clinical or pathological parameter such as tumor stage, estrogen receptor status, lymph node metastasis, and HER2 status. The sample size (36 tumors) is rather small, which restricts us from drawing a conclusion. We believe that a much larger cohort of patients is needed for testing whether miR-203 is also downregulated in metastatic primary breast cancers when compared to nonmetastatic cancers as in breast cancer cell lines examined and whether expression changes of miR-203 are associated with any clinicopathological findings.
Functionally, ectopic expression of miR-203 in metastatic breast cancer cell lines clearly suppressed cell invasion and motility (Fig. 4), further supporting a role of miR-203 in the suppression of tumor metastasis because motility and invasion are characteristic of metastatic cancer cells. This finding is consistent with previous studies in which expression of miR-203 suppressed cell invasion and bone metastasis in prostate cancer cells.14,25 On the other hand, ectopic expression of miR-203 also induced cell cycle arrest in MDA-MB-231 cells and apoptosis in BT549 cells (Fig. 3). Similar findings have been reported in other types of cancers. Ectopic expression of miR-203 promotes apoptosis and inhibits cell proliferation in bladder and esophageal cancer cells.26,27 It also induces differentiation by restricting proliferative potential of epidermal epithelial cells.2,3 Therefore, in addition to cell invasion, motility, and metastasis, miR-203 also appears to modulate cell proliferation and cell death in epithelial cells. However, it remains to be determined as to whether these distinct functions are intrinsically related. Although it is possible that miR-203 can modulate both cell proliferation and invasion/motility by targeting different genes, it is also possible that the induction of cell cycle arrest or cell death could contribute to the suppression of invasion and motility in cancer cells.
Different target genes have been reported for the function of miR-203 in different biological processes. For example, miR-203 targets ΔNp63, BMI1, and KLF4 to regulate cell proliferation and stemness,2,3,18,27 targets bcl-w to control cell death, 26 and targets a number of prometastatic genes including ZEB2, Bmi, Survivin, CKAP2, LASP1, BIRC5, WASF1, ASAP1, and Runx2 to restrict cell motility and metastasis.14,25 In this study, we identified SNAI2 as another target gene of miR-203, as its expression was downregulated by miR-203 at both the mRNA and protein levels, and mutation at either of the 2 potential targeting sequences abolished the effect of miR-203 on the expression of SNAI2 (Fig. 5). SNAI2 is a zinc-finger transcription factor important for cancer cells to downregulate epithelial markers and upregulate mesenchymal markers in order to become motile and invasive. 30 It initiates EMT in breast cancer cells. 31 Therefore, SNAI2 could well be one of the key molecules that are targeted by miR-203 in the control of cell motility, invasion, and metastasis.
The mechanism for the downregulation of miR-203 in metastatic breast cancer cell lines appears to be promoter methylation that silences gene transcription, as promoter methylation was obvious in each of the 3 metastatic breast cancer cell lines but not in nonmetastatic cell lines, and demethylating treatment restored miR-203 expression (Fig. 2). The same mechanism also occurs in other types of malignancies. For example, DNA hypermethylation appears to be responsible for miR-203 downregulation in squamous cell carcinomas, hepatocellular carcinoma, and hematological malignancies.8-11,32,33 Other than promoter methylation in the downregulation of miR-203 in cancer cells, how miR-203 is upregulated in primary breast cancer cells and whether and what other mechanisms are also responsible for the downregulation of miR-203 in metastatic cancer cells remain currently unknown.
We speculate that once breast cancer has formed, cells upregulate miR-203 as a self-defense mechanism to restrain them from invasion and metastasis, and downregulation of miR-203 in breast cancer cells by yet to be identified mechanisms contributes to more invasive and metastatic behavior of breast cancer. If true, evaluation of miR-203 expression in breast cancer could predict metastatic potential, and restoration of miR-203 expression could prevent metastasis of a tumor.
In summary, we found that miR-203 was upregulated in nonmetastatic but is downregulated in metastatic breast cancer cells, and promoter methylation appeared to mediate the downregulation. Expression of miR-203 induced cell cycle arrest and apoptosis and suppressed cell motility and invasion, and prometastatic SNAI2 was identified as a target gene of miR-203 in the control of cancer cell behavior. These findings suggest that miR-203 could modulate different characteristics of breast cancer cells and thus could be relevant to the detection and treatment of breast cancer.
Materials and Methods
Cell lines and primary tumor specimens
A total of 14 breast epithelial cell lines were used in this study, including 11 cancer cell lines (BT-474, BT549, CAMA-1, Hs578T, MCF7, MDA-MB-231, MDA-MB-361, MDA-MB-453, SKBR3, T-47D, ZR-75-1), 2 immortalized but nonneoplastic epithelial cell lines (184A1 and MCF10A), and 1 primary culture of human mammary epithelial cells (HMEC) (Cambrex, East Rutherford, NJ). Except for MDA-MB-453, which was purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (CBTCCCAS, Shanghai, China), all cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA) and were propagated as described previously. 34 Human embryonic kidney 293T cell line was purchased from CBTCCCAS and was cultured in DMEM supplemented with 10% FBS (Invitrogen, Shanghai, China) at 37°C and supplemented with 5% CO2 in a humidified chamber.
A total of 36 primary breast carcinoma tissues and their adjacent normal tissue specimens were obtained from surgically treated patients with breast cancer at the Cancer Hospital of Tianjin Medical University (Tianjin, China). Noncancerous tissues were harvested at least 5 cm from corresponding tumor sites, and surgical margins were confirmed to be clear of residual cancer. Use of the materials was approved by the hospital’s Ethics Review Committee. Tissues were cut into small pieces, snap frozen in liquid nitrogen, and stored in a –80°C freezer until use.
Genomic DNA and total RNA were extracted by using the DNeasy Tissue Kit (Qiagen, Shanghai, China) and the TRIzol reagent (Invitrogen, Beijing, China), respectively, according to the manufacturers’ manuals.
Plasmid construction
The pcDNA-pri-miR-203 expression vector contains pre-miR-203 with about 250-bp flanking sequences (total length: 637 bp; 85583452-85584088, GRCh37.p2), which was amplified by PCR from genomic DNA using the following 2 primers, 5′-CGGAATTCTGGCGGCTGGGATCCCCCAG-3′ (forward) and 5′-CCGCTCGAGCACCTCCCAGCAGCACTTGGCTCTC-3′ (reverse), which contain EcoRI or XhoI restriction sites (underlined) at their 5′ end for cloning. PCR products were digested with EcoRI and XhoI and cloned into the pcDNA3.1 (+) plasmid at EcoRI and XhoI sites. The 3′-UTR sequence (1029 bp, 56 nt from the start of 3′-UTR) of human SNAI2, which contains 2 putative miR-203 binding sites, was amplified by PCR using the following 2 primers, 5′-GACCGCGATCGCTGACAAATAAAGTCCAAAGGC-3′ (forward) and 5′-CT TAGTTTAAACAATCATGAAGCAAGTAAAGTCTC-3′ (reverse), which contain SgfI or PmeI restriction sites at their 5′ end. Three mutants of the 3′-UTR sequence, with mutation in either 1 of the 2 binding sites or in both binding sites, were generated by PCR using the Fast Mutagenesis System (TransGen Biotech, Beijing, China) using the following oligonucleotides: 5′-TTACATTGCTGCCAAATAGCCCAGA CTGAAAAGAACAGTAT-3′ (mutant 1, forward), 5′-ATA CTGTTCTTTTCAGTCTGGGCTATTTGGCAGCAATGTAA-3′ (mutant 1, reverse), 5′-TCATTAATGTACTTAA ACCGCCCAGAATGCATACCACAAATG-3′ (mutant 2, forward), and 5′-CATTTGTGGTATGCATTCTGGGCGGTTTAAGTACATTAATGA-3′ (mutant 2, reverse). PCR products were digested with SgfI and PmeI restriction enzymes and cloned into the 3′ end of the synthetic Renilla luciferase gene in the psiCHECK-2 vector at SgfI and PmeI sites. The sequences of inserted fragments were confirmed by DNA sequencing.
Regular and real-time RT-PCR
Regular RT-PCR was used to detect the expression of primary transcript and mature product of miR-203, SNAI2, E-cadherin, and vimentin, and real-time PCR was also used for the expression of mature miR-203. Briefly, for primary transcript, 11 µg of total RNA was reversely transcribed using oligo-dT primer (Takara, Tokyo, Japan), and 2 µL of the reverse transcription reaction mix was amplified by PCR with denaturation at 95°C for 2 minutes and 25 cycles at 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 1 minute. For mature product, 1 µg of total RNA was reversely transcribed using miR-203–specific stem-loop RT primer, and 2 µL of the reverse transcription mix was amplified by PCR with denaturation at 95°C for 2 minutes and 25 cycles (semiquantitative RT-PCR) or 50 cycles (quantitative real-time PCR) at 95°C for 10 seconds and 60°C for 1 minute. The average level of U6 was used as an internal control. Each data point was in triplicate. The SYBR green (Takara) method and the IQ5 Real-time PCR detection system (BioRad, Hercules, CA) were used for real-time PCR. Primer sequences were as follows: 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCTAGTG-3′ (stem-loop RT primer for miR-203), 5′-GTGCAGGGTCCGAGGT-3′ (real-time PCR, forward, miR-203), 5′-GCCGCGTGAAATGTTTAGG-3′ (real-time PCR, reverse, miR-203), 5′-CTCGCTTCGGCAGCACA-3′ (real-time PCR, forward, U6), 5′-AACGCTTCACGAATTTGCGT-3′ (real-time PCR, reverse, U6), 5′-TCTCCACTCACTGAGGCCTTAG-3′ (regular PCR, forward, pri-miR-203), 5′-TAGGTCCTTCACGAGTTTAGCG-3′ (regular PCR, reverse, pri-miR-203), 5′-TCACC CACACTGTGCCCATCTACGA-3′ (regular PCR, forward, β-actin), 5′-CAGCGGAACCGCTCATTGCCAATGG-3′ (regular PCR, reverse, β-actin), 5′-CAGGAACACAGGAGTCATCAGTGT-3′ (regular PCR, forward, E-cadherin), 5′-GAGGATTATCGTTGGTGTCAGTGA-3′ (regular PCR, reverse, E-cadherin), 5′- CCAAACTTTTCCTCCCTGA ACC-3′ (regular PCR, forward, vimentin), 5′- GTGATGCTGAGAAGTTTCGTTGA-3′ (regular PCR, reverse, vimentin), 5′- ATGAGGAATCTGGCTGCTGT-3′ (regular PCR, forward, SNAI2), 5′- CAGGAGAAAATGCCTTTGGA-3′ (regular PCR, reverse, SNAI2), 5′- TCGGAGTCAACGGATTTGGT-3′ (regular PCR, forward, GAPDH), and 5′- TTGGAGGGATCTCGCTCCT-3′ (regular PCR, reverse, GAPDH).
Methylation analysis
Genomic DNA was treated with bisulfite as previously described 35 and subjected to PCR using the same primers as previously published. 8 PCR products were cloned into the pMD18-T vector (Takara), and 3 clones were sequenced for each sample. For the demethylation analysis, breast cancer cell lines MDA-MB-231 and BT549 were cultured with or without 10 µM of 5-aza-2′-deoxycytidine (5-aza-dCyd) for 3 days, and the expression of miR-203 was detected by real-time PCR.
Cell proliferation assay
BT549 and MDA-MB-231 cells were seeded in 24-well plates at 1 × 105 cells per well with regular culture medium. After they were attached to plates, cells were washed and incubated with culture medium containing 5% FBS for 24 hours. Fifty nM of synthetic miRNA oligos (Ribobio, Guangzhou, China) mimicking miR-203 or nonspecific miRNA mimics (control) were transfected into cells using the Lipofectamine RNAiMAX reagent (Invitrogen). Culture media were renewed every other day. At 0, 2, 4, and 6 days after transfection, cells in one well were collected for RNA isolation, and cells in the remaining 3 wells were fixed with 10% trichloroacetic acid (TCA), stained with 0.4% sulforhodamine (SRB) (Sigma, St. Louis, MO), washed twice with 1% acetic acid, and subjected to the measurement of optical density in a spectrometer at 492 nm, as described in a previous study. 36
Cell cycle analysis
Cell cycle analysis was performed by using fluorescence-activated cell sorting (FACS) in a flow cytometer. Briefly, 1 × 106 cells were collected and fixed with 70% cold ethanol for 24 hours at –20°C, washed 3 times with PBS, and resuspended in PBS with 1 mg/mL RNase for 0.5 hours at 37°C. Cells were then stained with 0.025 mg/mL propidium iodide (PI) for 0.5 hours at 37°C in the dark. Analyses were carried out using the BD FACS Calibur flow cytometer (BD, Franklin Lakes, NJ) and the Cell Quest and ModFit computer program (BD).
Apoptosis assay
Apoptosis was measured by staining cells with Annexin V-FITC and PI and analysis with flow cytometry according to the manufacturer’s protocols (BD Pharmingen, San Diego, CA). Briefly, 1 × 106 cells transfected with miR-203 mimics or control mimics were washed in cold PBS and resuspended in 100 µL 1x staining buffer. After staining with Annexin V and PI (5 µL each) (BD Pharmingen) for 15 minutes at room temperature in the dark, cells were then subjected to flow cytometry sorting, and the data were analyzed by using the CellQuest computer program (BD).
Wound healing assay
Twenty-four hours after transfection into BT549 and MDA-MB-231 cells with miR-203 mimics or control mimics, some cells were used for expression confirmation with RT-PCR, while remaining cells were seeded in equal numbers into 6-well culture plates in RPMI 1640 medium supplemented with 1% FBS at about 95% confluence. Twelve hours after seeding, a vertical wound was created using a 10-µL pipette tip. Images were captured at designated times (0, 18, 24, and 36 hours) to assess the rate of gap closure.
Invasion assay
For invasion assays, BT549 and MDA-MB-231 cells transfected with miR-203 mimics or control mimics for 24 hours were collected in medium supplemented with 1% FBS and then either confirmed for miR-203 expression or plated in BD BioCoat BD Matrigel Invasion Chambers (BD China, Shanghai, China) at 1 × 105 cells per chamber. The membrane in the chamber was coated with Matrigel (BD China). Medium supplemented with 10% FBS was used in the lower chamber. Cells were incubated for 24 hours, and cells that did not invade through the pores of the membrane were scraped by a cotton swab. Cells on the lower surface of the membrane were fixed with polyoxymethylene (Sigma) and stained with 0.1% crystal violet (Sigma) for 0.5 hours. Stained cells were counted under a microscope in 4 randomly selected fields, and the average was used to indicate cell invasion.
Luciferase reporter assay
Expression plasmids of reporter vectors containing SNAI2 3′-UTR or its mutants were transiently transfected into 293T cells, along with miR-203 or control mimics, using the Lipofectamine 2000 reagent (Invitrogen) following the manufacturer’s manual. Forty-eight hours after transfection, cells were lysed with 100 µL of Passive Lysis Buffer (Promega, Fitchburg, WI), and luciferase levels were measured from 20 µL of lysate using the dual luciferase reporter assay on the Berthold FB12 luminometer (Berthold, Bad Wildbad, Germany) following a previously described procedure. 37 Changes in expression of Renilla luciferase were normalized by the firefly luciferase activities.
Western blotting
Western blotting was done using a previously published procedure. 38 Rabbit polyclonal SNAI2 antibody was purchased from BD Biosciences (Franklin Lakes, NJ) and was used with a dilution of 1:800. Mouse monoclonal β-actin antibody was from Santa Cruz Biotechnology (Santa Cruz, CA) and used with a dilution of 1:3,000.
Footnotes
Acknowledgements
The authors thank Jianping Jenny Ni for her comments on the preparation of the article.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This work was supported by the National Nature Science Foundation of China [grant numbers 30870980, 31171250, and 30625032]; National “863” Program of China [grant number 2006AA02A249]; National “973” Program of China [grant number 2009CB521700]; Doctoral Fund of Ministry of Education of China [grant number 200800551032]; Key Projects of National Science and Technology [grant number 2009ZX08009-408151B]; and National Cancer Institute of the National Institutes of Health [grant number R01CA085560].