
Abstract
The Cas family proteins, p130Cas (Cas) and NEDD9, are adaptor molecules that regulate cytoskeletal dynamics to promote multiple cellular processes, including migration, invasion, proliferation, and survival. Because these functions are also critical for tumor initiation, growth, and metastasis, Cas and NEDD9 are well positioned to contribute to these oncogenic processes. Indeed, mouse models of cancer show that these proteins function during multiple stages of disease progression. Furthermore, in many human cancers, high expression of Cas and NEDD9 is associated with advanced stage disease and is predictive of poor outcome. This review explores the contribution of Cas and NEDD9 during cellular transformation and neoplastic growth, tumor progression, metastasis, and the development of therapeutic resistance. Given these roles, Cas and NEDD9 may prove to be viable candidates for use as biomarkers and therapeutic targets.
p130Cas/BCAR1 (p130 Crk-associated substrate/breast cancer antiestrogen resistance 1; Cas, hereafter), one of a four-member family of protein scaffold and adaptor molecules, plays a central role in transducing signals from cell surface receptors and cytoplasmic protein tyrosine kinases (PTKs). Cas has no catalytic activity but instead exerts its influence on cell behavior by functioning as a platform for the assembly of multiprotein complexes. Cas is a substrate for many PTKs, most notably the non–receptor tyrosine kinases c-Src (Src) and focal adhesion kinase (FAK).1-3 Progressive tyrosine phosphorylation of Cas drives its interaction with a number of binding partners whose spatial and temporal localization contribute to basic cellular processes such as adhesion, migration and invasion, proliferation and survival, morphogenesis, and homeostasis.4,5 The full integration and manifestation of these signals require structural reorganization of the actin/microtubule cytoskeleton, and one major function of Cas is to promote the assembly of the protein machinery capable of affecting such change.6,7
Because Cas is an important component of signaling networks that regulate a variety of somatic cell behaviors, it is not surprising that its aberrant expression and activity have been implicated in human disease. 7 An association between Cas and cancer has existed since its discovery as a mediator of cell transformation by the v-Src and v-Crk oncoproteins 8 ; subsequent work has shown that Cas expression contributes to tumorigenesis in mice 9 and humans.5,7 Research links alterations in Cas activity and expression to many cancers, including those of the breast, prostate, colon, lung, and brain and several hematopoietic malignancies. 7 High Cas expression is associated with high tumor grade, advanced-stage disease, decreased relapse-free survival, and/or increased mortality in patients with breast, ovarian, prostate, and non-small-cell lung cancer.10-13
The evolution of cancer is driven by the deregulation of signaling pathways that are responsible for maintaining normal cell behaviors. Thus, a protein such as Cas, which is an essential component of numerous signal transduction networks, is ideally positioned to contribute to cell transformation and disease progression. This review focuses on the functions of Cas and its family member NEDD9 and explores them in the context of (1) cell transformation and neoplastic growth, (2) tumor progression, and (3) metastasis. Although these steps represent a continuum of the oncogenic process, we have chosen to address each independently to provide a framework for understanding Cas/NEDD9 function in cancer. We end by discussing a possible role for Cas and NEDD9 in the establishment of resistance to several classes of anticancer drugs and their potential value as prognostic markers and therapeutic targets.
The Cas Family
Cas family members
In addition to Cas, the Cas family is comprised of NEDD9 (neural precursor cell expressed, developmentally down-regulated 9, also known as HEF1 or Cas-L), EFS (embryonal Fyn-associated substrate, also known as Sin), and CASS4 (Cas scaffolding protein family member 4, or HEPL). While Cas is an abundant, ubiquitously expressed protein, expression of the other members is, to varying degrees, temporally regulated and tissue-specific (for a more complete review of the differential expression patterns of the Cas family, see Tikhmyanova et al. 7 ). Cas and NEDD9 are the most extensively studied of this family, and both are implicated in the development and progression of cancer. Consequently, we have chosen to focus this review on these two family members.
Basic structure of Cas/NEDD9
As Cas proteins have been the topic of several excellent reviews,5-7,9,14 we provide only a brief overview of the basic structure of Cas and NEDD9. These proteins contain four conserved domains: an amino-terminal Src homology 3 (SH3) domain; a large substrate domain that contains multiple YxxP sequences that, when phosphorylated, provide docking sites for Src homology 2 (SH2) domain-containing proteins; a serine-rich four-helix bundle; and a highly conserved carboxy-terminal domain that contains binding sites for the Src family of protein tyrosine kinases and a second four-helix bundle that promotes hetero- and homo-dimerization (Fig. 1).
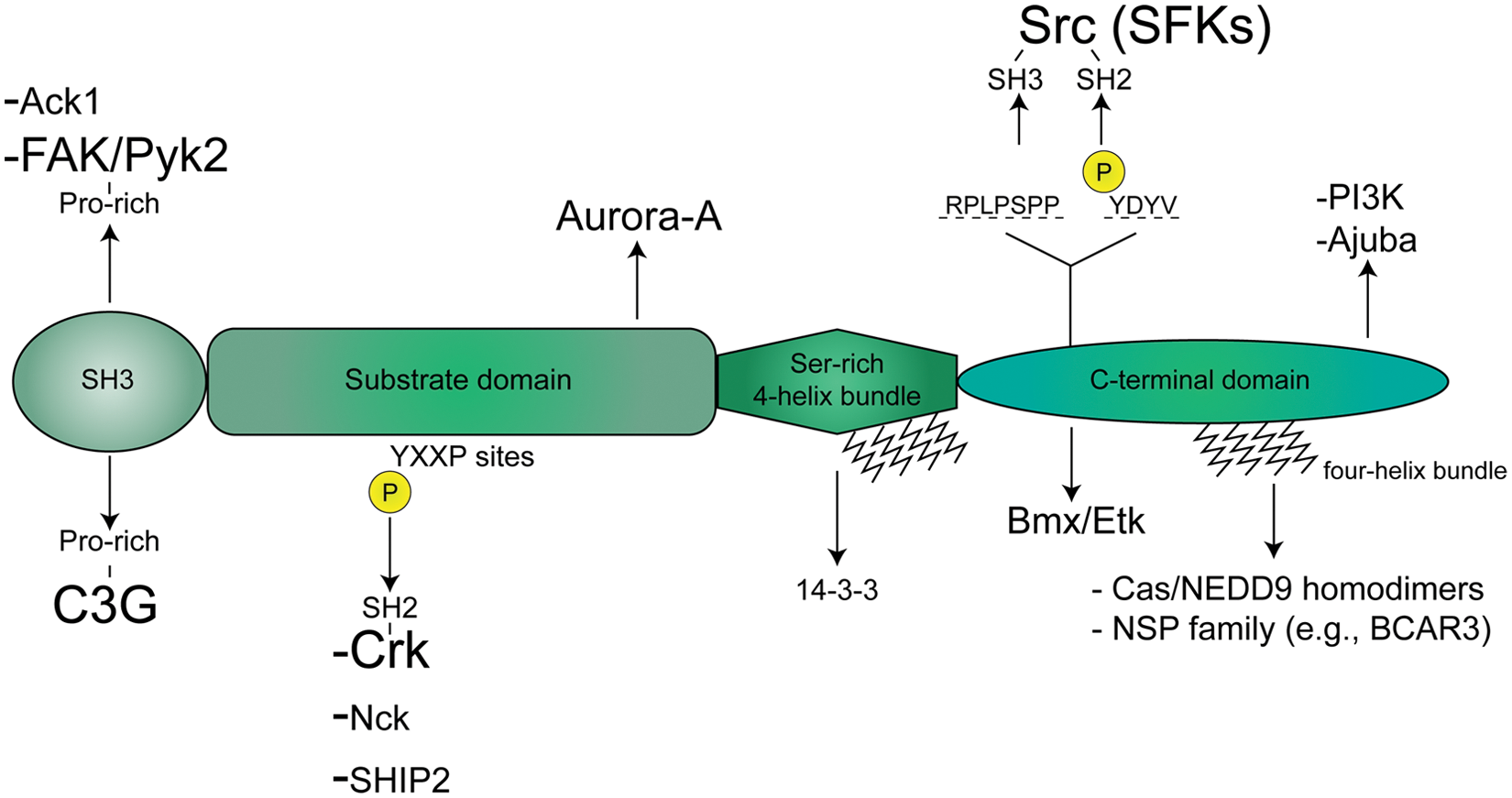
Cas/NEDD9 domain structure and binding partners. Cas and NEDD9 share four conserved domains that mediate their associations with other proteins: an N-terminal SH3 domain, a substrate domain containing multiple YxxP motifs that are phosphorylated by Src family kinases (SFKs), a serine-rich four-helix bundle, and a C-terminal domain that contains binding sites for SFKs, other Cas family members, and members of the novel SH2-containing protein (NSP) family. For binding partners highlighted in this review (FAK/Pyk2, C3G, Crk, Nck, and Src), specific domains of interaction are shown.
Cellular functions and binding partners of Cas and NEDD9
Cas and NEDD9 transduce signals downstream of integrin engagement and receptor tyrosine kinase (RTK) activation by establishing physical interactions with a number of cytoplasmic proteins (Fig. 1). While numerous binding partners of Cas and NEDD9 have been identified, three proteins have been most consistently implicated in Cas/NEDD9 activity and signaling: the PTKs Src and FAK, and the SH2 domain-containing adaptor molecule Crk. These proteins bind to Cas/NEDD9 through the carboxy-terminal domain, the SH3 domain, and the substrate domain, respectively (Fig. 1). 6
Cas participates in the transduction of chemical and mechanical signals that are initiated by ligand activation of growth factor, cytokine, and integrin receptors. One of the more extensively studied Cas signaling pathways is activated upon integrin engagement; NEDD9 has been shown to function through similar mechanisms. 7 Upon adhesion to the cell matrix, Cas forms a complex with Src and FAK. 3 The interaction of Cas with Src stabilizes the kinase in a conformationally active state by relieving autoinhibitory intramolecular interactions.15,16 Interestingly, Cas overexpression drives Cas-Src association, resulting in the tyrosine phosphorylation of multiple Src substrates, including Cas and FAK.15,17 In this way, Cas overexpression can prolong and amplify Src activity in the absence of exogenous stimuli.
Phosphorylation of Cas within its substrate domain results in the creation of binding sites for Crk and other small adaptors such as Nck.1,15,18 Cas-Crk complexes activate Rho and Ras family GTPases such as Rac1 and Rap1 via the recruitment of two guanine nucleotide exchange factors (GEFs), DOCK180 and C3G.18-21 These GTPases regulate cytoskeletal dynamics and normal cellular processes such as motility, proliferation, and cell division but can also contribute to tumorigenesis.22,23 The work reviewed below provides evidence that Cas/NEDD9-cytoskeletal signaling is involved in the initiation, maintenance, and progression of tumorigenesis.
Cell Transformation and Neoplastic Growth
Mouse models
A number of mouse models of breast cancer reveal a role for Cas and NEDD9 in hyperplasia, transformation, tumor initiation, and malignant growth.5,9,24,25 Several investigators have examined the requirement for Cas or NEDD9 in oncogene-driven breast cancer models. While global Cas deletion is embryonic lethal, 4 NEDD9 knockout mice (NEDD9–/–) are viable and fertile. 24 The formation of neoplastic lesions is reduced and early tumor development is impaired in MMTV-PyVmT/NEDD9–/– mice compared with MMTV-PyVmT/NEDD9+/+ mice. 24 These differences in tumor development are coincident with reduced Src and FAK activity in NEDD9–/– tumors. 24 Similarly, small interfering RNA (siRNA)–mediated knockdown of Cas significantly decreases tumor formation in both orthotopic and spontaneous mouse models of ErbB2-mediated mammary tumorigenesis. 5 A reduction in Src activity is observed in Cas-depleted tissues, as is a decrease in FAK phosphorylation at tyrosine 925 (Y925), a site that has been shown to be a Src substrate and involved in the angiogenic switch seen in many solid tumors.5,26 These data underscore an essential role for Cas/NEDD9-scaffolding activity and PTK activation in oncogene-driven transformation and tumor establishment.
While not sufficient to promote tumor formation, transgenic overexpression of Cas in the mammary glands of mice accelerates gland development and induces highly disorganized epithelial structures and hyperplastic growth. 9 Increased cell proliferation correlates with an increase in Src activity in this hyperplastic mammary tissue. Interestingly, these Cas transgenic mice also display a decrease in apoptosis in ductal epithelial cells during involution. 9 Finally, double transgenic mice that co-overexpress Cas and HER2/Neu under the control of the MMTV promoter exhibit accelerated tumor formation compared with animals that express HER2/Neu alone, demonstrating the ability of Cas to augment the transforming capacity of HER2/Neu in the mammary epithelium. 9
Xenograft models further establish a role for Cas signaling in transducing proliferative and survival signals that promote malignant growth. Depletion of Cas from the mouse mammary tumor cell line 4T1 reduces tumor cell proliferation and inhibits primary tumor growth following mammary fat pad engraftment. 25 Similarly, while loss of Cas in a transforming growth factor-β (TGF-β)–driven model of mammary tumorigenesis has no effect on tumor establishment, it does significantly reduce tumor outgrowth. 25 This is hypothesized to be due to the ability of Cas to induce resistance to TGF- β-mediated growth inhibition by binding to Smad2/3, thus preventing their phosphorylation and the subsequent induction of apoptosis, cytostasis, and/or homeostasis.25,27 In support of Cas mediating cell survival, increased apoptosis is observed in carcinoma cells isolated from HER2/Neu-transgenic mice following Cas knockdown. 9
Anchorage independence and morphological changes
In culture, anchorage-independent growth and changes in cell morphology are metrics often used to define cellular transformation. Cas plays an essential role in cellular transformation mediated by Src, anaplastic lymphoma kinase (ALK), and ErbB2 (Fig. 2A).4,5,28 Following expression of these proto-oncogenes, Cas-null fibroblasts are unable to form colonies in soft agar and exhibit differences in morphology due to defects in actin cytoskeletal organization and structure.4,5,28 For example, Cas-deficient cells that express a transforming mutant of Src exhibit a defect in actin filament aggregation at invadopodia, a structural hallmark of transformation.4,29
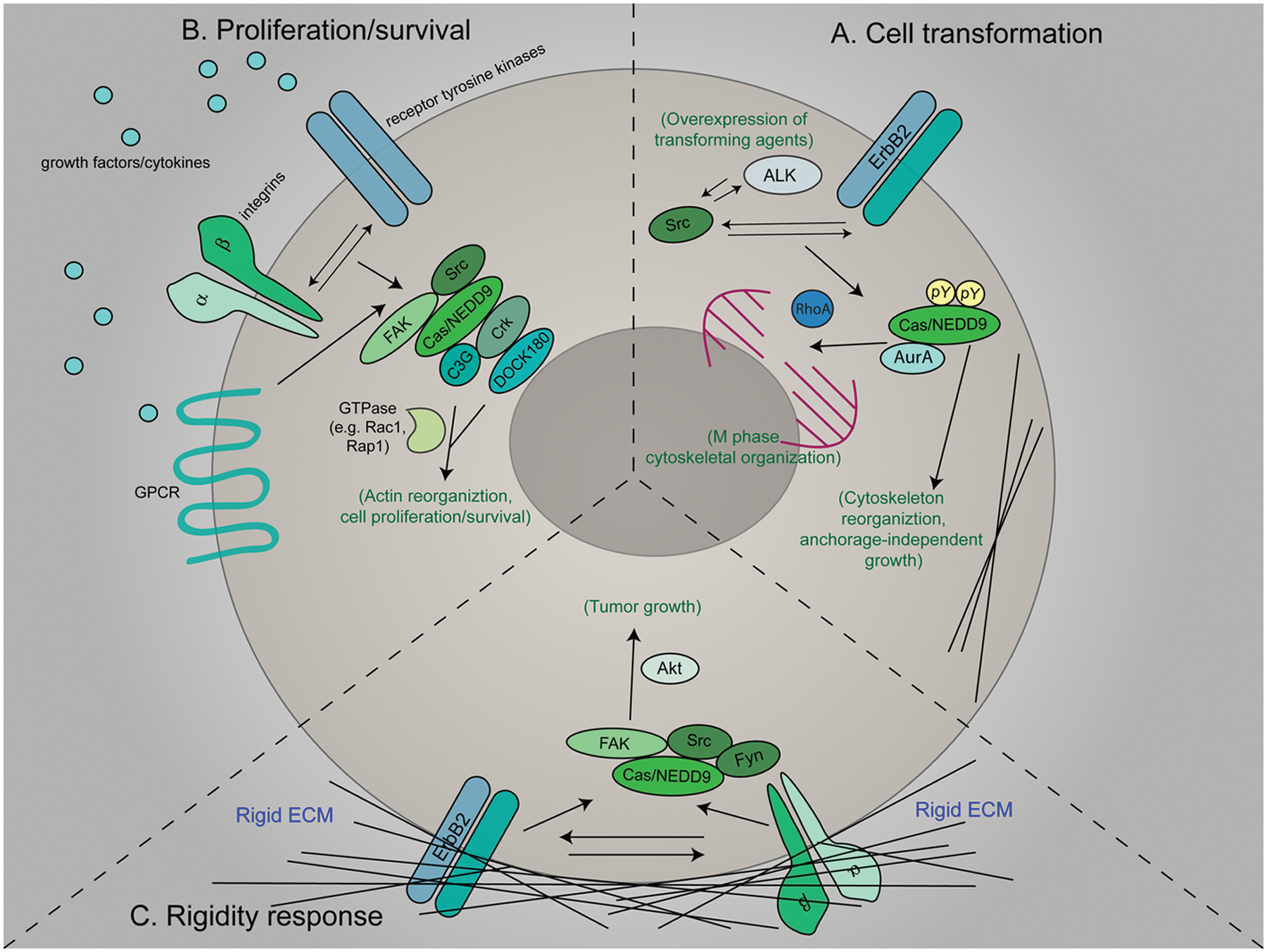
Cas/NEDD9 signaling pathways involved in cell transformation and tumor growth. (
Conservation of genomic stability
The precise spatial and temporal regulation of proteins that control mitotic cell division is important for maintaining genomic stability. NEDD9 expression increases during G2/M and is required for the cytoskeletal reorganization that occurs as cells undergo mitosis (Fig. 2A). 30 In breast cancer cells, NEDD9 binds to and positively regulates the Aurora-A (AurA) kinase, a molecule that initiates mitotic entry through the activation of cyclinB/Cdk1.30-32 Upon overexpression of NEDD9, multipolar spindles and supernumerary centrosomes are observed, similar to what is seen following overexpression or constitutive activation of AurA. 30 NEDD9 overexpression is also accompanied by a delay in mitotic progression and a failure to complete cytokinesis. 33 Conversely, knockdown of NEDD9 results in poorly formed mitotic spindles and an increase in the percentage of cells that fail to form cleavage furrows and enter M phase.30,33 It has been shown that NEDD9 controls mitotic cytoskeletal organization by regulating RhoA activity through interactions with the RhoA GEF ECT2. 33 Given the essential role that NEDD9 plays during mitosis, it is reasonable to postulate that deregulated or aberrant expression of this molecule contributes to the genomic instability that is associated with tumor initiation and growth.
Unlike NEDD9, Cas protein levels remain constant through the cell cycle. However, Cas and FAK become heavily phosphorylated on serine and dephosphorylated on tyrosine residues at the onset of mitosis.34,35 These changes correlate temporally with the breakdown of Cas/Src/FAK signaling complexes, the disassembly of focal adhesions and actin stress fibers, and the acquisition of a rounded morphology. 34 Given the increases in Src activity and Cas/FAK phosphorylation observed in cells overexpressing Cas, 15 the regulation of Cas phosphorylation during mitotic entry may become disrupted upon Cas overexpression. This could in turn adversely affect cytoskeletal dynamics during cell division, leading to genomic instability.
Tumor Progression
Within the tumor microenvironment, communication between tumor cells and the associated stroma is essential for progression to a more invasive state. 36 Cas and NEDD9 can contribute to this process through (1) the propagation of growth factor and cytokine signaling cascades in response to factors within the tumor microenvironment; (2) the promotion and regulation of angiogenesis to create new vasculature; (3) remodeling of the surrounding tissue architecture to further expose the tumor to growth-sustaining agents and peptides; and (4) translation of external physical forces into growth-promoting cues. In each case, Cas/NEDD9-dependent modulation of the cytoskeleton may play important roles in both tumor and stromal cells to mediate these processes.
Growth factor signaling
Cas/NEDD9 signaling networks are activated in response to a number of signaling molecules found within the tumor microenvironment. Cas (and in many cases, NEDD9) becomes phosphorylated following stimulation with epidermal growth factor (EGF), 37 vascular endothelial growth factor (VEGF),38,39 TGF-β,25,40 insulin-like growth factor-1 (IGF-1), 41 hepatocyte growth factor (HGF), 42 bombesin, lysophosphatidic acid (LPA), and platelet-derived growth factor (PDGF). 43 An intact cytoskeleton is required for Cas phosphorylation following growth factor stimulation, as treatment with cytochalasin D abolishes the observed increase in tyrosine phosphorylation in response to EGF, bombesin, and PDGF.37,43 Similarly, cytochalasin D blocks tyrosine as well as serine/threonine phosphorylation of NEDD9 following exposure to TGF-β. 40 Given the dependency of Cas and NEDD9 phosphorylation on an intact cytoskeleton and the concomitant increase in phosphorylation and cytoskeletal remodeling in response to many of these growth factors,37,44 a functional relationship between Cas/NEDD9 signaling complexes and the cytoskeleton appears to be critical for integrating growth factor input during tumor growth (Fig. 2B).
Upon phosphorylation, Cas signaling complexes are generated that transduce mitogenic and survival signals. For example, treatment of cells with EGF induces Cas-Crk coupling and a reorganization of the cytoskeleton. 37 Cas-Crk signaling is also required for cell survival in response to EGF, insulin, and serum. 45 Expression of a dominant-negative Rac1 molecule blocks this effect, demonstrating a role for Cas/Crk/Rac1 in mitogen-induced cell survival. 45 Finally, expression of a dominant-negative Cas mutant lacking the substrate domain inhibits PDGF-driven cell proliferation by blocking FAK signaling to JNK. 46 These data support a central role for Cas in a wide array of mitogenic signaling pathways that can function both within the tumor cells and in the supporting cells of the tumor microenvironment.
Angiogenesis
To increase growth-sustaining factors in the tumor microenvironment, tumor cells must create new blood vessels. 47 The tumor vasculature is generated by endothelial cell polarization and migration toward angiogenic factors such as VEGF. 48 Endothelial cell polarity and migration during new vessel sprouting require the activity of Rac1, Cdc42, and RhoA, which have been shown to be activated downstream of Cas or NEDD9.19,33,48,49 Human vascular endothelial cell (HUVEC) migration toward VEGF requires Cas expression and phosphorylation by the FAK family member, proline-rich tyrosine kinase 2 (Pyk2). 42 Pyk2-dependent Cas phosphorylation, cytoskeletal reorganization, and cell migration are also observed in brain endothelial cells upon VEGF treatment. 38 Interestingly, Pyk2 kinase activity regulates the expression of Cas and NEDD9 in pulmonary vein endothelial cells (PVECs), as the overexpression of a catalytically inactive mutant of Pyk2 dramatically reduces the cellular levels of both proteins. The expression of this Pyk2 mutant also abolishes the ability of PVECs to form capillary-like structures in response to treatment with the angiogenic factor phorbol 12-myristate 13-acetate (PMA). 50 Together, these findings suggest that Cas and NEDD9 act downstream of Pyk2 (and possibly other PTKs) to regulate the cytoskeletal reorganization necessary for endothelial cell polarization and migration during angiogenesis (Fig. 3A).
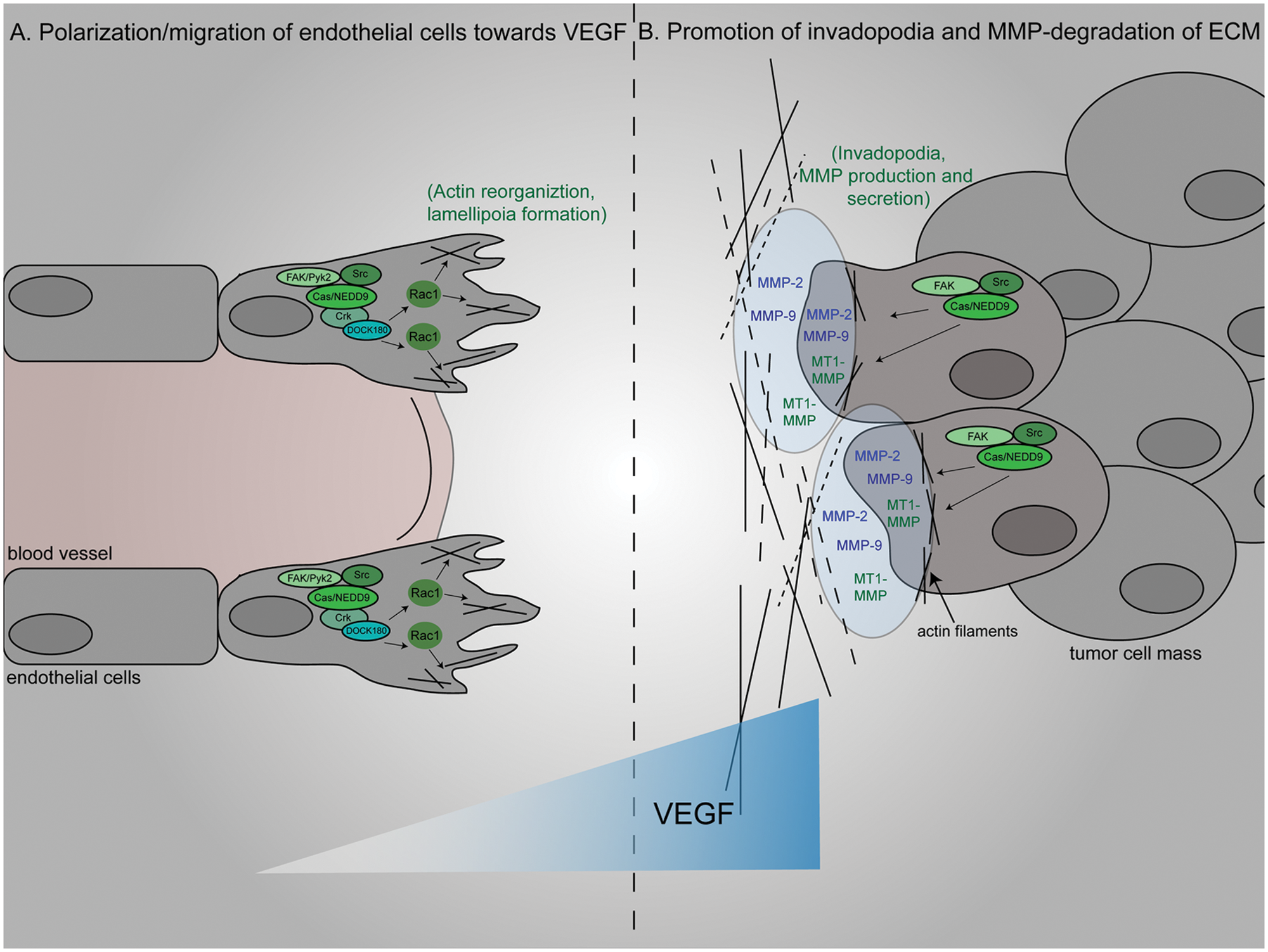
Proposed model of tumor vascularization regulated by Cas/NEDD9 signaling complexes. (
ECM degradation
Degradation of the tumor ECM further increases access to growth factors and cytokines. This is accomplished via secretion of proteases by both tumor cells and other cells in the tumor microenvironment. Parallel with the induction of Cas-dependent endothelial cell migration, VEGF also stimulates expression of the matrix-degrading factors matrix metalloproteinase (MMP)-2 and -9 and MT1-MMP in a NEDD9-dependent manner in the oral squamous cell carcinoma cell line SCC-9. 39 Similarly, in Src-transformed fibroblasts and fibrosarcoma cells, Cas participates in Src/FAK signaling cascades to facilitate the expression, activity, and secretion of these same MMPs.17,51,52 Cas-FAK interactions are required for the accumulation of these proteases at actin-rich invadopodia, 52 which are structural adaptations of the cytoskeleton that are associated with cancer cell aggressiveness. 29 Thus, Cas/NEDD9 signaling complexes are assembled in response to the local accumulation of VEGF and facilitate Src/FAK-dependent ECM degradation through the formation of MMP-containing invadopodia (Fig. 3B).
Mechanotransduction
In addition to soluble factors, the tumor microenvironment provides mechanical signals from the ECM that control local tumor growth. In many cases, tumors are more rigid than normal tissue, and this increased stiffness can regulate tumor cell growth, morphology, motility, invadopodia, and degradation of the ECM.53-56 Cas is a well-defined mechanosensor that responds to changes in ECM rigidity, stretching, and stress.21,57,58 Interestingly, the growth profiles of many cancer cell types show a preference for growth on increasingly rigid substrates. 56 Cas signaling is activated in cells cultured on stiff matrices and is required to restore rigidity-dependent spreading and growth mediated by the Src family kinase Fyn.54,57 Proliferation is increased in colorectal cancer cells grown in three-dimensional collagen matrices pretreated with lysyl oxidase (LOX) to induce matrix stiffening. 59 This enhancement in proliferation is dependent on the kinase activities of Src and FAK. 59 Ectopic expression of LOX in MCF7 breast cancer cells similarly induces Src and FAK activity and the formation of the Cas/Crk/DOCK180-signaling complex.60,61 Interestingly, tumors grown in mammary fat pads of mice that have elevated levels of LOX are significantly larger than those grown under control conditions. 55 This coincides with increased levels of Cas protein and FAK activity in the tumors. These data indicate that Cas/Src/FAK signaling complexes may function as mechanosensors that contribute to tumor growth through their ability to integrate changes in extracellular mechanical signals (Fig. 2C).
Metastasis
Mouse models
In vivo models implicate Cas and NEDD9 signaling complexes during metastasis. For example, lung metastases are significantly reduced following tail vain injection of Cas-depleted ErbB2-overexpressing breast cancer cells compared with control counterparts. 5 Similarly, pulmonary dissemination of Cas-deficient cells is significantly impaired in a transgenic model of TGF-β-driven breast cancer. 25 Finally, Cas phosphorylation has been shown to be an essential event during Rap1-dependent, EGFR-induced pancreatic cancer cell metastasis. 62
A comparative genomic analysis of mouse melanoma models and human melanoma tissue establishes NEDD9 as a “metastasis gene.” 63 In support of this finding, the MMTV-PyVmT/NEDD9–/– mouse displays a trend toward decreased lung metastases. 24 Together, these results support a role for Cas/NEDD9 in the dissemination and subsequent colonization of tumor cells to foreign tissues.
Initiation of EMT
Prior to metastasis, it has been proposed that tumor cells undergo “epithelial-mesenchymal transition” (EMT), a process characterized by functional, genetic, and phenotypic changes coincident with cytoskeletal reorganization and loss of cell-cell and cell-matrix interactions.47,64 Cas and NEDD9 are implicated in the initiation of EMT (Fig. 4A). 65 Overexpression of either Cas or NEDD9 in MCF7 breast cancer cells leads to Src-dependent lysosomal degradation of the epithelial marker E-cadherin, and their co-overexpression accelerates this process. 65 Additionally, overexpression of NEDD9 drives EMT in breast cancer cells by concurrently increasing the expression of mesenchymal markers and decreasing the expression of epithelial markers through the promotion of Snail and Slug protein levels, two transcription factors that regulate EMT. 66 The initiation of EMT can be triggered by the signaling molecules TGF-β, EGF, and Wnt,64,66 all of which signal downstream to Cas or NEDD9.37,40,67 Furthermore, Wnt signaling induces the expression of Twist, another transcription factor that regulates EMT.68,69 Twist, in turn, positively controls the expression of NEDD9 and DOCK3, a DOCK180-related GEF, and mediates cell invasion and mesenchymal movement in head and neck squamous cell carcinoma cell lines through the promotion of NEDD9/DOCK3-dependent Rac1 activation. 69
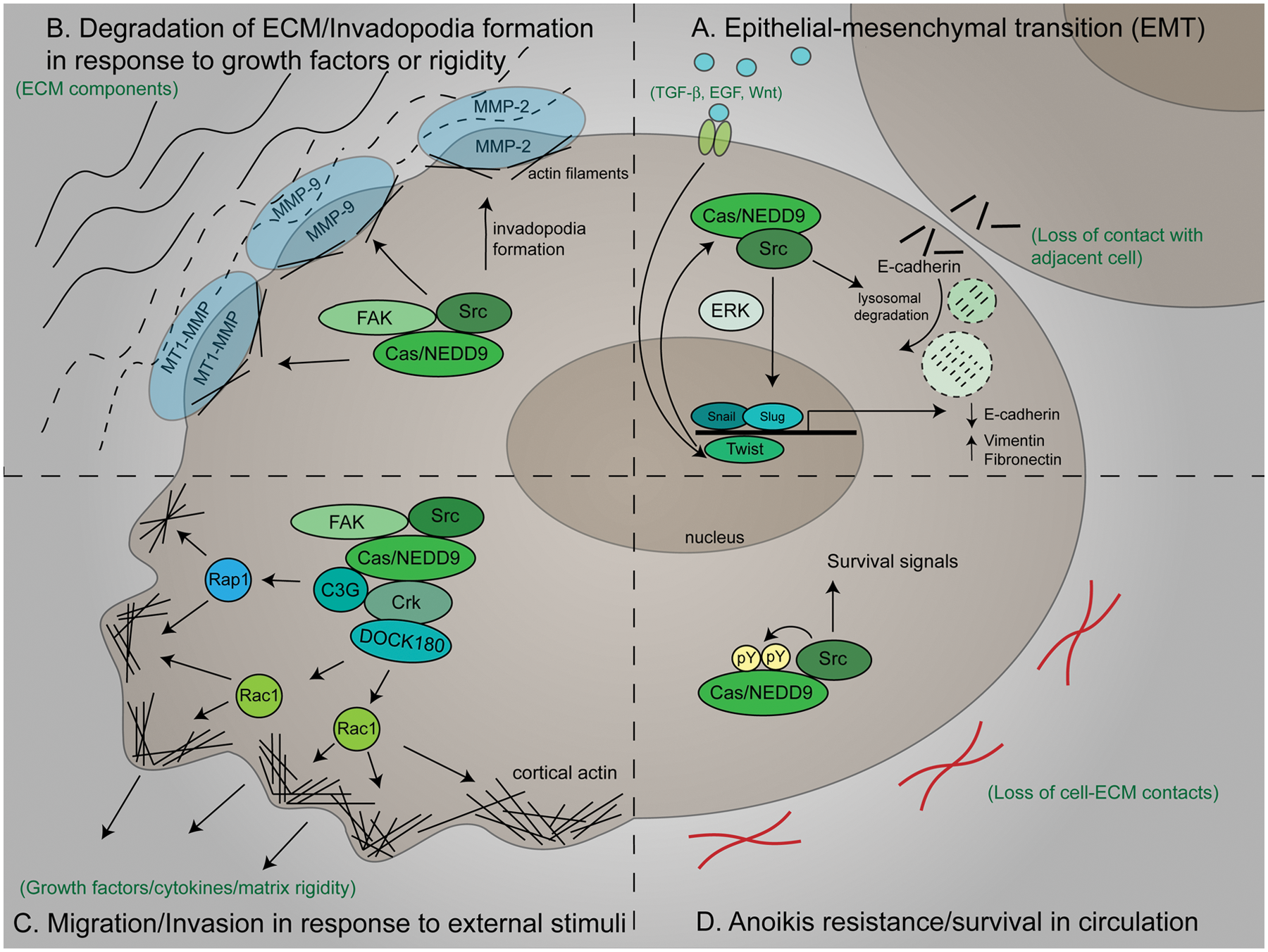
Cas/NEDD9 signaling complexes function in cellular processes during metastatic progression. (
Control of cell migration and invasion
An extensive body of literature describes the control of migratory and invasive cell behaviors through Cas/NEDD9-dependent cytoskeletal remodeling (see Tikhmyanova et al. 7 ). Cas or NEDD9 overexpression increases the migratory and invasive potential of cells in in vitro systems.17,20,70 Three-dimensional organotypic cultures also provide evidence that Cas promotes invasive cell behavior, as Cas overexpression causes acinar filling and the formation of invasive multiacinar structures in mouse and human mammary epithelial cultures.25,71
The regulation of cell migration and invasion by Cas and NEDD9 is initiated via phosphorylation of the substrate domain by Src and FAK (Fig. 4B and 4C). This induces the assembly of a multiprotein complex containing small adaptor proteins (Crk or Nck) and GEFs (DOCK180/DOCK3 or C3G). Depending on the cell type and/or stimulus, this complex then activates Rac1 or Rap1 to induce the cytoskeletal remodeling and membrane protrusions that facilitate cell movement.14,20,45,49,62,70,72-75 This signaling pathway plays an essential role in mediating movement toward growth factors and in translating mechanical forces into biochemical signals that initiate motility. Similarly Bmx/Etk, another non–receptor tyrosine kinase, uses a Cas/Crk signaling pathway to regulate haptotactic cell migration. 76
Cas and NEDD9 may also influence cell migration through interactions with the Zyxin family of LIM proteins, which localize to focal adhesions and the actin cytoskeleton during migration and in response to mechanical stretch to reinforce and remodel actin filaments.7,77,78 Ajuba, a Zyxin family member, associates with Cas and localizes it to the leading edge of migrating cells, resulting in Rac1 activation and lamellipodia extension. 79 Additionally, recent data indicate that zyxin and Cas cooperate to ensure efficient cell migration in three-dimensional cultures. 80
Intravasation of tumor cells into the cardiovascular and lymphatic systems requires invasion through the basement membrane and vessel walls. Invadopodia formation and protease activity play key roles in intravasation, and as discussed in the preceding section, Cas/NEDD9 signaling is important for both of these processes (see Fig. 3B). Cas overexpression markedly increases the invasive capacity of Src-transformed cells by enhancing invadopodia formation and MMP-2 activity. 17 Additionally, through its ability to promote actin cytoskeletal remodeling, invadopodia formation, and MMP-9 activity via Rac1 and JNK activation, the Cas/Src/FAK signaling complex acts as a positive regulator of invasion toward both EGF and serum. 51 Cas/FAK complexes also promote pancreatic tumor cell invasion by inducing MT1-MMP degradation at focal adhesions. 52 Similarly, NEDD9 co-localizes with MT1-MMP at invadopodia and induces invadopodia formation, matrix degradation, and cell invasion. 39
Cas has been shown to promote tumor cell invasion in response to mechanical stimulation (Fig. 4B and 4C). Cas becomes phosphorylated and relocalized to invadopodia in response to increased ECM rigidity. 54 In addition, Cas overexpression further amplifies the invadopodia-associated, matrix-degrading activity of breast cancer cells plated on rigid substrates. 54 Taken together, these studies implicate the activity of Cas/NEDD9 signaling complexes in the promotion of tumor cell behaviors necessary for invasion through tissue and intravasation into blood and lymphatic vessels.
Survival and adhesion in the vasculature
Once in the circulation, the majority of tumor cells die. 81 The inhibition of anoikis, a cell death program activated following cell-ECM detachment, is critical in mediating the survival of metastatic tumor cells in circulation. 82 In anoikis-resistant lung cancer cells, Cas is highly phosphorylated in suspension (Fig. 4D). 82 Inhibition of Src activity or the expression of a dominant-negative Cas molecule lacking the substrate domain abolishes Cas phosphorylation and restores the anoikis response.
As tumor cells travel through the circulation, they become trapped in the capillary beds of distant organs and adhere to endothelial cells or areas of exposed basement membrane. 81 Extravasation into the secondary tissue requires an invasive process through the basement membrane that is similar to what is described above (see Fig. 4B and 4C). Blood and lymph flow through vessels creates a mechanical force known as shear stress, which can stimulate cancer cell adhesion and Src/Cas/Crk/DOCK180/Rac1 signaling.83-87 Interestingly, phosphorylated Cas and Rac1 activity is localized to the area of the cell that lies downstream of flow. 87 Actin protrusions and cell movement are similarly oriented in this direction. Concurrently, Cas signaling is deactivated in regions of the cell facing flow; these regions are characterized by large focal adhesions and prominent stress fibers. 87 Through this spatial regulation, Cas is positioned to promote extravasation of adhered tumor cells into surrounding tissues.
Cas/NEDD9 in Drug Resistance, Clinical Outcome, and Therapeutic Potential
The gene breast cancer antiestrogen resistance 1 (BCAR1) was identified more than a decade ago through a retroviral-insertion mutagenesis screen designed to identify genetic factors that cause breast cancer cells to become resistant to the antiestrogen drug tamoxifen. 88 Further analysis identified the BCAR1 gene product as the human homologue of murine Cas. 89 Ectopic expression of Cas in the estrogen-dependent ZR-75-1 cell line, which expresses low levels of endogenous Cas, allows for proliferation in the presence of the antiestrogens tamoxifen and ICI 182,780. 89 Subsequent studies have shown that Cas induces tamoxifen resistance through a mechanism that requires Cas-Src association and Src PTK activity. The establishment of this resistance mechanism also involves the activity of two Src substrates, EGF receptor (EGFR) and signal transducer and activator of transcription (STAT) 5b. 90 Cas overexpression also protects MCF7 breast cancer cells from the cytotoxic effects of adriamycin, and this protective effect is abolished upon inhibition of Src activity. Conversely, depletion of Cas renders more resistant breast cancer cell lines sensitive to this agent. 91
NEDD9 is implicated in the development of therapeutic resistance. NEDD9 protein levels are increased in imatinib-resistant gastrointestinal stromal tumor cells. 92 NEDD9 depletion is sufficient to restore sensitivity, as is inhibition of Src kinase activity. 92 Based on these data, overexpression of Cas or NEDD9 appears to establish a Src-dependent pathway that bypasses the intended cytotoxic effects of these drugs to sustain cell viability and growth.
Evaluation of Cas and NEDD9 in human tumors reveals that elevated expression of these proteins is an indicator of tumor stage and grade, disease progression, and poor outcome.10-13,63 Relapse-free and overall survival times inversely correlate with Cas protein levels in a large series of breast cancer tissue samples. 10 Additionally, analysis of microarray data from 295 early-stage breast cancer biopsies shows that high levels of Cas correlate with high ErbB2 expression. 71 Moreover, those tumors that co-overexpress Cas and ErbB2 are associated with increased metastasis and shorter overall survival times. 71 A correlation between NEDD9 overexpression and metastasis is similarly observed in human melanoma. 63 In ovarian carcinomas, Cas overexpression is associated with advanced-stage disease, decreased relapse-free survival, and increased mortality. 11 In prostate cancer, high-grade tumors exhibit increased Cas expression compared with their low-grade counterparts. 12 In addition, high Cas expression is predictive of relapse in well-differentiated prostate tumors. 12 Finally, elevated Cas expression in a cohort of non-small-cell lung cancer specimens correlates with poorly differentiated tumors and decreased overall survival. 13 While further validation is needed, these findings suggest that Cas and NEDD9 expression may prove to be viable markers of tumor aggressiveness and predictors of poor disease outcome.
Recently, the therapeutic efficacy of targeting Cas has been examined in preclinical models. The in vivo delivery of nanoliposomes containing Cas-specific siRNAs in combination with docetaxel significantly reduces tumor burden in mice bearing taxane-sensitive ovarian tumors. 11 Cas knockdown also restores sensitivity of taxane-resistant tumors to docetaxel, an important finding as many human tumors develop resistance to these drugs. 11
Summary and Perspectives
Cas and NEDD9 are integral to a number of essential cellular processes. However, when viewed in a broader context, these individual processes can be considered components of a larger signaling network that actively regulates cytoskeletal dynamics to integrate cell proliferation, survival, and motility during tumorigenesis. For example, tumor cells coordinate survival and migratory pathways to invade through tissue, survive in the circulation, and colonize secondary sites during the establishment of metastatic lesions.45,81 The in vitro and in vivo studies reviewed here demonstrate that Cas and NEDD9 are necessary components of metastatic pathways.
The preclinical research that links Cas and NEDD9 to tumor initiation, progression, and metastasis is consistent with clinical data that demonstrate that elevated expression of these proteins is associated with disease progression and mortality.10-13,63 However, further studies are needed to better understand the physiological functions of these proteins during tumorigenesis. The ability to attribute distinct functions to these proteins during the promotion of specific stages of tumor development will be useful in assessing the value of these Cas family members as clinically significant biomarkers and therapeutic targets.
Therapies that target Cas and NEDD9 are particularly intriguing because these proteins control the behavior of tumor cells as well as other cells within the tumor microenvironment. While these drugs may prove to be successful as single agents, their greatest efficacy may be derived in combination with drugs that target the kinase activities of Src, FAK, or HER2, as the functions of Cas and NEDD9 are intimately linked to these kinases. The potential therapeutic benefit of the proposed combinatorial therapies awaits further investigation into whether high Cas/NEDD9 expression correlates with increased activity or expression of Src, FAK, or HER2 in human tumors.
Footnotes
Acknowledgements
We thank members of the laboratory for valuable input into this review.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We acknowledge grant support from the NIH (R01 CA096846 to AHB, CA40042 and 1U54 GM64346 to JTP, F31 CA130168 to MSG) and the UVA Cancer Center through the Women’s Oncology Research Fund, the Charlottesville Women’s 4-Miler Breast Cancer Research Fund, and the NCI Cancer Center Support Grant (P30 CA44579).