
Abstract
Ect2 is a member of the human Dbl family of guanine nucleotide exchange factors (RhoGEFs) that serve as activators of Rho family small GTPases. Although Ect2 is one of at least 25 RhoGEFs that can activate the RhoA small GTPase, cell culture studies using established cell lines determined that Ect2 is essential for mammalian cell cytokinesis and proliferation. To address the function of Ect2 in normal mammalian development, we performed gene targeting to generate Ect2 knockout mice. The heterozygous Ect2 +/ – mice showed normal development and life span, indicating that Ect2 haplodeficiency was not deleterious for development or growth. In contrast, Ect2 – / – embryos were not found at birth or postimplantation stages. Ect2 – / – blastocysts were recovered at embryonic day 3.5 but did not give rise to viable outgrowths in culture, indicating that Ect2 is required for peri-implantation development. To further assess the importance of Ect2 in normal cell physiology, we isolated primary fibroblasts from Ect2 fl/fl embryos (MEFs) and ablated Ect2 using adenoviral delivery of Cre recombinase. We observed a significant increase in multinucleated cells and accumulation of cells in G2/M phase, consistent with a role for Ect2 in cytokinesis. Ect2 deficiency also caused enlargement of the cytoplasm and impaired cell migration. Finally, although Ect2-dependent activation of RhoA has been implicated in cytokinesis, Ect2 can also activate Rac1 and Cdc42 to cause growth transformation. Surprisingly, ectopic expression of constitutively activated RhoA, Rac1, or Cdc42, known substrates of Ect2, failed to phenocopy Ect2 and did not rescue the defect in cytokinesis caused by loss of Ect2. In summary, our results establish the unique role of Ect2 in development and normal cell proliferation.
Introduction
Rho family small GTPases are regulators of diverse cellular processes that include cell proliferation and survival, actin organization and cell shape, polarity and movement, and endocytosis and exocytosis.1,2 There are 20 human Rho GTPases, with RhoA, Rac1, and Cdc42 as the best-characterized members. Rho GTPases function as GDP-GTP–regulated binary switches that are activated in response to extracellular stimuli. Activated Rho GTPases in turn associate with effectors that then stimulate cytoplasmic signaling networks. 3 Rho-specific guanine nucleotide exchange factors (RhoGEFs) promote GDP-GTP exchange and formation of active Rho-GTP, 4 whereas Rho-specific GTPase activating proteins (RhoGAPs) accelerate hydrolysis of the bound GTP to stimulate formation of inactive Rho-GDP. 5
There are 83 human RhoGEFs, with Dbl family RhoGEFs comprising the largest group (69 members).4,6 Dbl RhoGEFs are characterized by a conserved Dbl homology (DH) catalytic domain and an adjacent C-terminal pleckstrin homology (PH) regulatory domain. The DH domain may be highly or broadly specific for a subset of Rho GTPases. For example, Tiam1 is a specific activator of Rac, and Asef is specific for Cdc42, whereas Vav is broadly active and can activate RhoA, Rac, Cdc42, and RhoG.
The significantly greater number of RhoGEFs relative to their GTPase substrates suggests highly redundant mechanisms for Rho GTPase activation. This apparent redundancy in activators is particularly striking for RhoA, where there are at least 25 Dbl RhoGEFs that can activate this Rho GTPase. 4 However, the multiple RhoGEFs for one Rho GTPase provide mechanisms where a specific Rho GTPase can be activated by divergent signals, leading to distinct spatiotemporal patterns of activation and signaling. This divergence in RhoGEF regulation and function is reflected by the otherwise highly divergent sequences that flank the DH-PH domains. 3 RhoGEFs are generally large proteins (>1,000 amino acids) and often contain several domains involved in their localization, association with other proteins, regulation of GEF catalytic activity, and Rho GTPase effector selectivity. For example, among all human RhoGEFs, only Ect2 contains tandem BRCT (BRCA1 C-terminal) domains that are normally found in proteins (e.g., BRCA1, TP53BP1) involved in DNA damage signaling, repair, and coordination of cell cycle checkpoints.7,8 The BRCT domains may serve to regulate the RhoGEF catalytic activity and additionally can interact with phosphorylated proteins that dictate Ect2 subcellular localization.9-11
Ect2 is also only one of 2 Dbl family RhoGEFs, aside from Net1, 12 that exhibits a nuclear localization in interphase cells. 13 Because Rho GTPases are membrane associated and/or cytoplasmic, it has been proposed that this nuclear localization is an inactive repository for Ect2 since disruption of nuclear localization activates Ect2 transforming activity. 14 However, a recent study found that nuclear Ect2 may be active. 15 Thus, whether nuclear Ect2 has a function during interphase and distinct from cytokinesis remains unresolved. The best-known function of Ect2 involves its requirement for cytokinesis in vitro.16,17 In early mitosis (prometaphase) when the nuclear envelope is dissolved, Ect2 is released into the cytoplasm. As cells enter anaphase, Ect2 is recruited to the central spindle by the central spindlin protein complex comprised of Cyk4/MgcRacGAP and MKLP1/CHO1. The central spindle facilitates contractile ring formation, leading to cleavage furrow ingression and midbody formation. Ect2 is recruited to the midbody where Ect2 then activates RhoA to facilitate midbody abscission and completion of cytokinesis. Ectopic expression of a noncatalytic fragment of Ect2 that includes the BRCT domains caused inhibition of U-2 OS human osteosarcoma 13 or adenovirus-transformed HEK 293T human kidney neuronal-like 18 to complete cytokinesis, resulting in the accumulation of multinucleated cells. Similarly, microinjection of anti-Ect2 antibody into HeLa human cervical carcinoma cells also inhibited cytokinesis. 13 Subsequent studies using transient RNA interference suppression of Ect2 expression in HEK 293T 18 or HeLa10,19 cells also demonstrated a requirement for Ect2 in cytokinesis in vitro. No assessment of mammalian Ect2 function in normal cells in vitro or in vivo has been described.
Although Ect2 was identified originally as an activated oncoprotein that was activated by truncation and loss of N-terminal sequences that include the BRCT domains and nuclear localization signals, 20 no such truncated proteins have been detected in human cancers. Instead, recent studies have identified aberrant overexpression and mislocalization of full-length Ect2 to the cytoplasm in glioblastoma and lung cancer tumor tissue and cell lines.21-23 These studies used RNA interference to suppress Ect2 expression, which caused impaired lung tumor cell anchorage-independent growth and Matrigel (BD Biosciences, Franklin Lakes, NJ) invasion in vitro and reduced tumorigenic growth in vivo. Interestingly, in lung tumor cells, Ect2 suppression did not impair cytokinesis, indicating that Ect2 function in oncogenesis is distinct from that in cytokinesis. 23 Further support for this possibility was provided by the observation that constitutively activated Rac1 could rescue the loss of endogenous Ect2 and restore lung tumor cell growth. 23 This finding contrasts with previous studies that found that RhoA is the substrate critical for Ect2-dependent cytokinesis.16,17
Previous studies of mammalian Ect2 function have been done in established cell line studies in vitro. To evaluate the function of Ect2 in vivo, in normal cells, and in the context of heterogenous tissue, we generated both floxed (conditional) and Δfloxed (constitutive) knockout mice to assess the consequences of Ect2 loss in development and normal cell proliferation. Constitutive loss of Ect2 causes embryonic lethality, with defective growth at the late blastocyst stage. Utilizing mouse embryo fibroblasts derived from Ect2 fl/fl conditional mice, we determined that loss of Ect2 completely impaired cell proliferation and migration in vitro, causing accumulation of cells in G2/M phase and formation of enlarged multinucleated cells. Surprisingly, expression of activated Rho GTPases failed to rescue the loss of Ect2 to restore cell proliferation or migration. Our observations provide further evidence for the highly unique function of this RhoGEF in normal cell physiology.
Results
Generation of Ect2 mutant mice
To evaluate the function of Ect2 in mammalian development, we generated a conditional null mutation of the mouse Ect2 locus by gene targeting. Our targeting strategy focused on exon 8, which encodes the N-terminal tandem BRCT domains, by flanking it with loxP sites (Fig. 1A). Cre-mediated deletion resulted in a Δfloxed allele lacking exon 8 that is a null mutation based on the complete loss of Ect2 protein expression that we describe below. Figure 1A shows a schematic diagram of the wild-type Ect2 allele, the targeting vector, the targeted allele after homologous recombination, and the resulting floxed (conditional) and Δfloxed (conditional null) alleles.
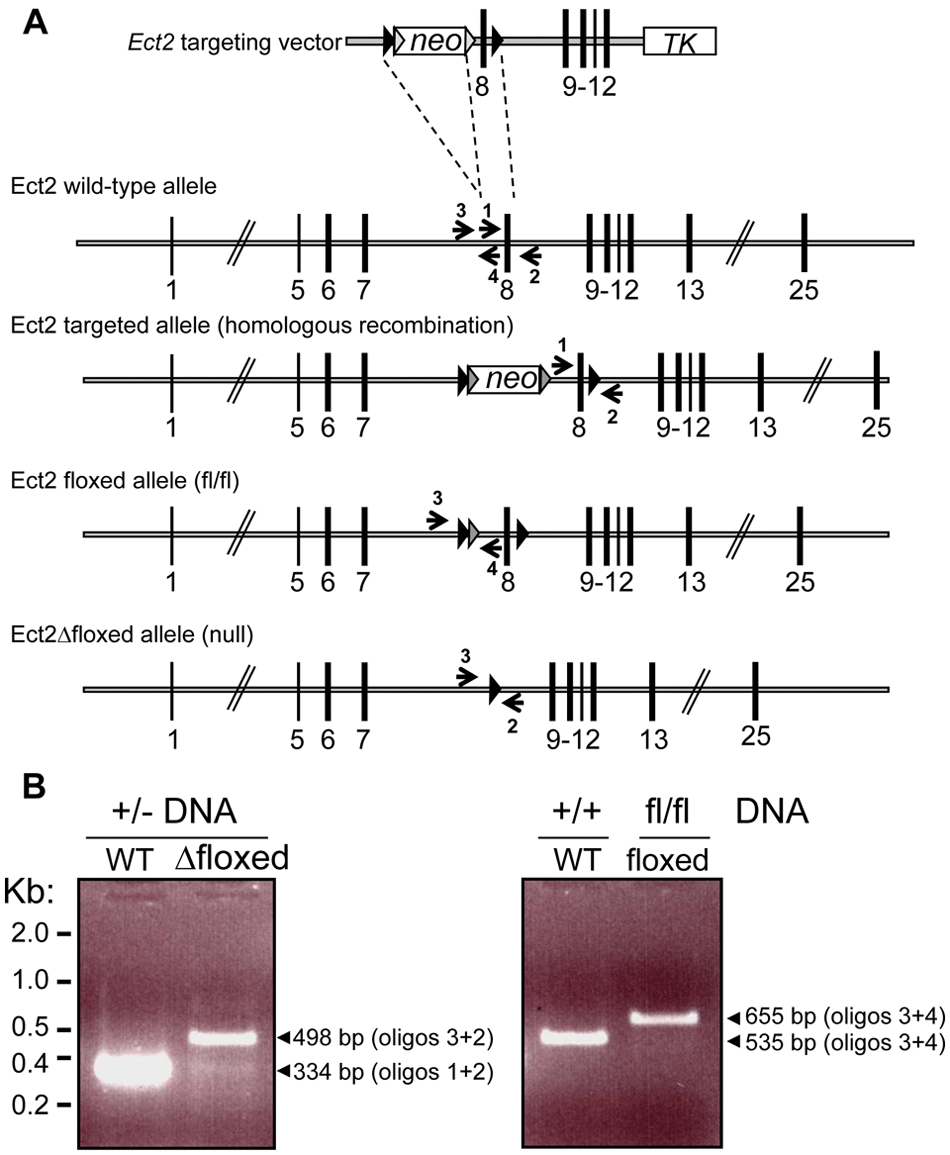
Targeted disruption of the mouse Ect2 gene produces a null mutation. (
Peri-implantation lethality of Ect2 mutant embryos
A ubiquitous Cre driver was used to generate constitutive null heterozygotes. Intercrosses of these Ect2 +/− mice failed to produce any homozygous mutants at birth among 224 offspring, suggesting that Ect2 −/− is embryonic lethal (Table 1). Therefore, we performed Ect2 +/− intercrosses as timed matings and genotyped embryos at progressively earlier stages of development. No Ect2 −/− embryos were obtained at embryonic days (E) 15.5, 13.5, or 8.5 (Table 1). These results suggested that Ect2 is required for either preimplantation or early postimplantation development.
Genotypes of Weanlings and Embryos from Ect2 + /– Intercrosses

Examination of maternal uteri exhibited empty deciduas, suggesting peri-implantation lethality.
To investigate the peri-implantation stage in more detail, we performed additional Ect2 +/− intercrosses, isolated blastocysts at E3.5, and analyzed their outgrowth ex vivo. 24 Homozygous Ect2 −/− blastocysts had a morphologically normal trophectoderm (TE) and inner cell mass (ICM) (Fig. 2). We cultured 125 blastocysts for 5 days and then scored the resulting outgrowths and determined their genotypes. After this culture period, 84% of wild-type outgrowths and 92% of Ect2 +/− outgrowths were scored as normal based on the presence of both TE and extensive ICM (Fig. 2 and Table 2). In contrast, no Ect2 −/− outgrowths were scored as normal. Instead, 14% of homozygous blastocysts failed to hatch from their zona pellucida, and the remaining 86% were scored as abnormal based on the ICM not being viable. These results indicate Ect2 is required for peri-implantation development.
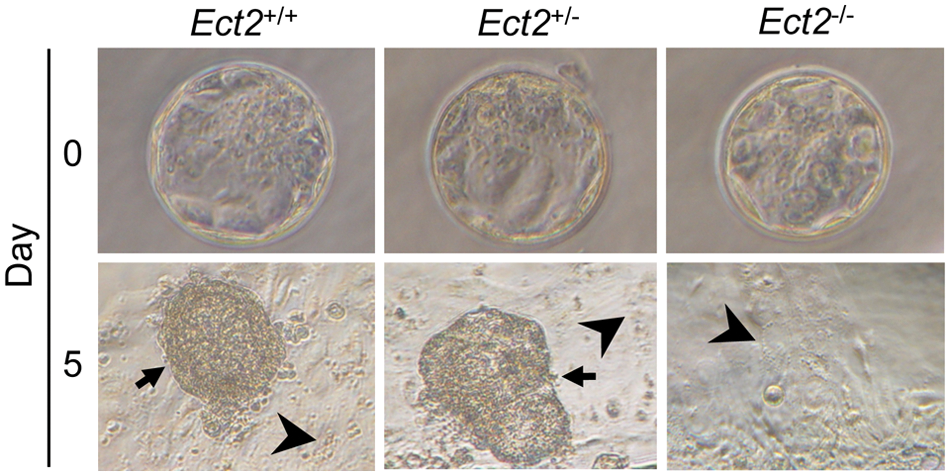
Ect2 −/− blastocysts display abnormal growth in vitro. Photographs of wild-type (+/+), heterozygous (+/−), and homozygous null (−/−) blastocysts at the time of isolation at E3.5 (day 0) and after being cultured for 5 days (day 5). Arrows point to inner cell mass, which is missing in Ect2 −/− blastocysts. Arrowheads point to trophectoderm.
Phenotype and Genotype of Blastocyst Outgrowths Derived from Ect2 Intercrosses

Survival and good outgrowth for trophectoderm and inner cell mass.
Hatched, but inner cell mass failed to grow.
Normal development of Ect2+/− mice
Because Ect2 −/− mice were embryonic lethal, we monitored Ect2 +/− mice for haploinsufficiency. We compared Ect2 +/− mice to their wild-type littermates at several stages of development. No changes in weight, gross appearance, or histology were found in Ect2 +/− compared to wild-type littermates at E15.5, 30 days, and 1 year (Fig. 3A and data not shown). The life span of Ect2 +/− mutant mice was then followed longitudinally. No differences in survival were observed between Ect2 +/− mice and their wild-type littermates (Fig. 3B).
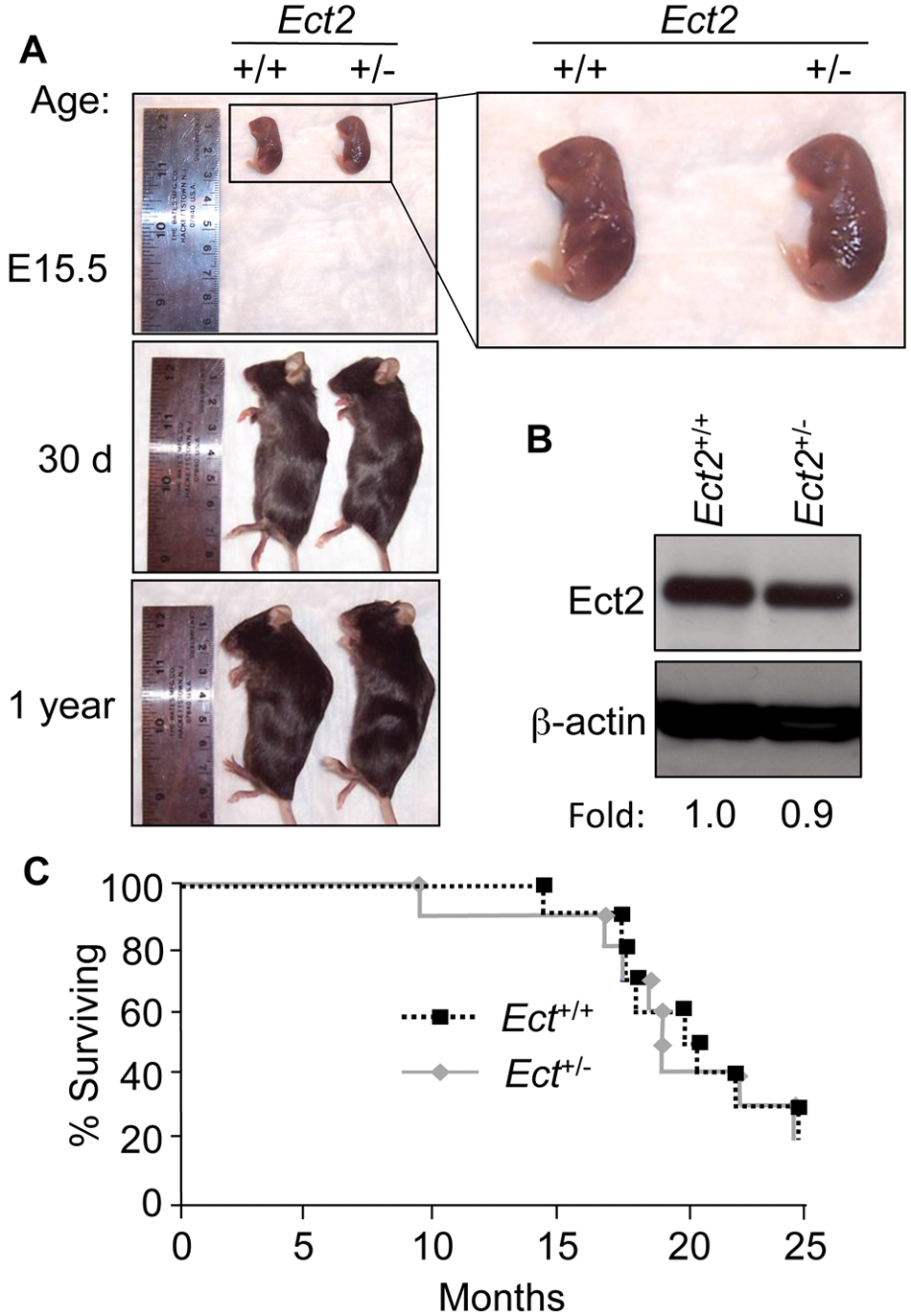
Ect2
+/−
mice exhibit normal development and survival. (
Given that we did not identify phenotypic differences between Ect2 +/− mice and their wild-type littermates, we examined whether the reduced gene dosage of Ect2 +/− heterozygotes resulted in corresponding decreases in the level of Ect2 protein expression. We isolated mouse embryo fibroblasts (MEFs) at E15.5 from both Ect2 +/+ and Ect2 +/− mice and examined Ect2 protein levels (Fig. 3C). We found no significant differences in Ect2 protein expression between Ect2 +/ – and Ect2 +/+ MEFs, suggesting that Ect2 +/− mice had equivalent levels of Ect2 protein compared to their wild-type littermates.
Ect2 deficiency results in multinucleated cells with altered morphology and impaired migration
As Ect2 deficiency was embryonic lethal, we isolated and immortalized MEFs from Ect2 fl/fl and Ect2 +/+ mice. These cells were then infected with recombinant adenovirus expressing Cre (Ad-Cre) to excise exon 8 of Ect2. An infection efficiency of 90% to 100% was achieved, and this resulted in a complete loss of Ect2 expression (Fig. 4A). Ad-Cre–infected Ect2 fl/ fl MEFs were much larger in size than their wild-type counterparts and appeared to have abundant nuclei and cytoplasm at 72 hours after infection. To examine this observation further, we sparsely plated Ad-Cre–infected MEFs on coverslips 24 hours after infection and allowed the cells to grow for 72 hours. Using fluorescent microscopy, we found that Ad-Cre–infected Ect2 fl/fl MEFs were large and multinucleated. Both Ect2 fl/fl and Ect2 +/+ MEFs appeared normal when infected with a control recombinant adenovirus expressing green fluorescent protein (Ad-GFP) (Fig. 4B and data not shown). We quantitated the difference in nucleation and found that Ad-Cre–infected MEFs, which are Ect2-deficient, had a significantly higher percentage of multinucleated cells (~85%) compared to wild-type MEFs (<3%), suggesting that loss of Ect2 expression causes a defect in mitosis (Fig. 4C). To further assess Ect2 expression in Ect2 fl/f l MEFs infected with Ad-Cre, immunofluorescence of Ect2 was performed, and the Ect2 fl/ fl MEFs infected with Ad-Cre showed a loss of specific Ect2 staining, whereas the nontreated control showed clear Ect2 nuclear staining as expected (Fig. 4D).
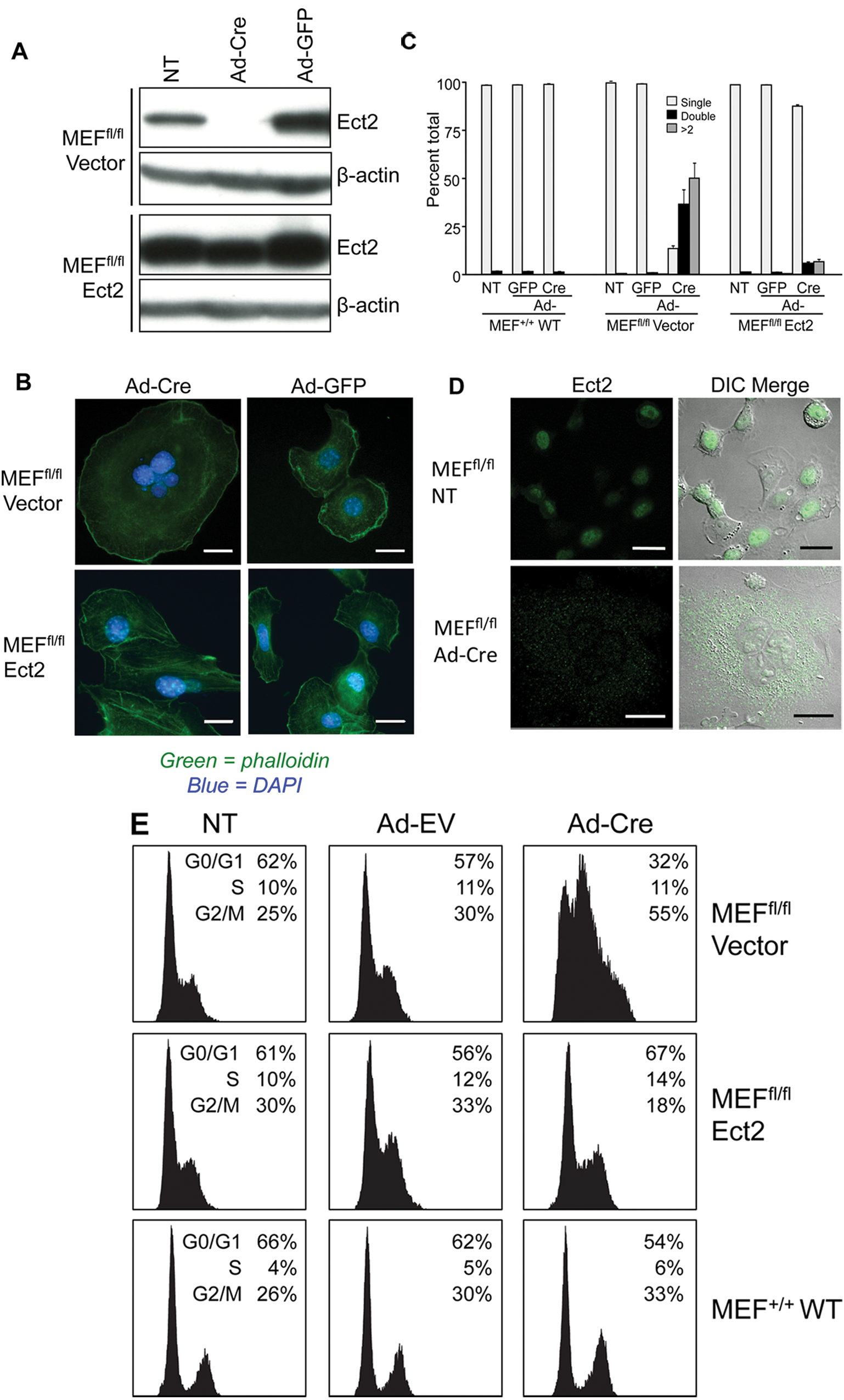
Ect2
fl/fl
MEFs deficient in Ect2 expression exhibit accumulation of multinucleated cells and can be rescued with ectopic expression of Ect2. MEFs were isolated from Ect2
fl/fl
and Ect2
+/+
mice and were infected with recombinant adenovirus expressing Cre recombinase (Ad-Cre) to ablate Ect2 expression. Ectopic expression of Ect2 was achieved by infection with the FUGW lentivirus expression vector encoding human Ect2, with control cells infected with the empty FUGW vector. (
The increase in multinucleation associated with loss of Ect2 expression was also quantified by cell cycle analysis, and the Ect2 fl/ fl MEFs showed a large increase in cells in G2/M phase when infected with Ad-Cre as compared to the Ad-GFP control virus and untreated MEFs (Fig. 4E). Furthermore, Ad-Cre infection had no effect on wild-type MEFs, indicating the specificity of the phenotype. Finally, the defect in cytokinesis could be rescued by infection with the FUGW lentivirus expression vector that encodes full-length wild-type human Ect2, demonstrating that impaired cytokinesis was due to loss of Ect2 expression (Fig. 4).
Because Ect2 is an activator of Rho GTPases and Rho GTPases regulate actin reorganization, we also evaluated the consequences of Ect2 loss on actin organization. Phalloidin staining of control and Ad-Cre–infected MEFs showed similar low levels of actin stress fibers, but there was a reduction in membrane ruffling (Fig. 4B). Because Rho GTPases can regulate cell attachment and migration, we also utilized time-lapse microscopy to monitor cell motility. Control Ad-GFP–infected MEFs exhibited high motility, whereas Cre-mediated loss of Ect2 caused a striking near-complete loss of this activity (Suppl. Movie S1).
Rho GTPases cannot rescue the multinucleated phenotype caused by loss of Ect2
There is considerable evidence that suggests that the requirement for Ect2 during cytokinesis is for activation of RhoA at the central spindle and midbody to complete mitosis.16,17 However, there is also evidence that Ect2 activation of Cdc42 but not Rac1 is important for mitosis. 19 In contrast, a recent study determined that ectopic expression of constitutively activated Rac1 but not RhoA (or Cdc42) rescued the loss of endogenous Ect2 to restore lung carcinoma cell anchorage-independent growth and invasion. 23 Thus, the specific Rho GTPase important for the role of Ect2 in cytokinesis remains to be elucidated.
To determine if a specific activated Rho GTPase could rescue the cytokinesis defect caused by the ablation of Ect2 expression, hemagglutinin (HA) epitope-tagged wild-type or constitutively active GTPase-deficient mutants (RhoA(Q63L), Rac1(Q61L), and Cdc42(Q61L)) were each stably expressed in Ect2 fl/f l MEFs prior to Cre expression (Fig. 5A). Surprisingly, expression of wild-type or constitutively active GTPases failed to rescue the multinucleation phenotype associated with the loss of Ect2 expression (Fig. 5B). Because fast cycling mutants with enhanced intrinsic nucleotide exchange activities may better model GTPase activation by a RhoGEF, 25 we also ectopically expressed RhoA(F30L), Rac1(F28L), or Cdc42(F28L) in Ect2-depleted MEFs (Suppl. Fig. S1B). No rescue of cytokinesis was seen. These negative results contrast with the ability of constitutively activated Rac1(Q61L) to rescue loss of cytoplasmic mislocalized Ect2 in lung cancer cells 23 and may indicate the requirement for a more precise temporal and spatial control of GTPase activity during normal cell cytokinesis.
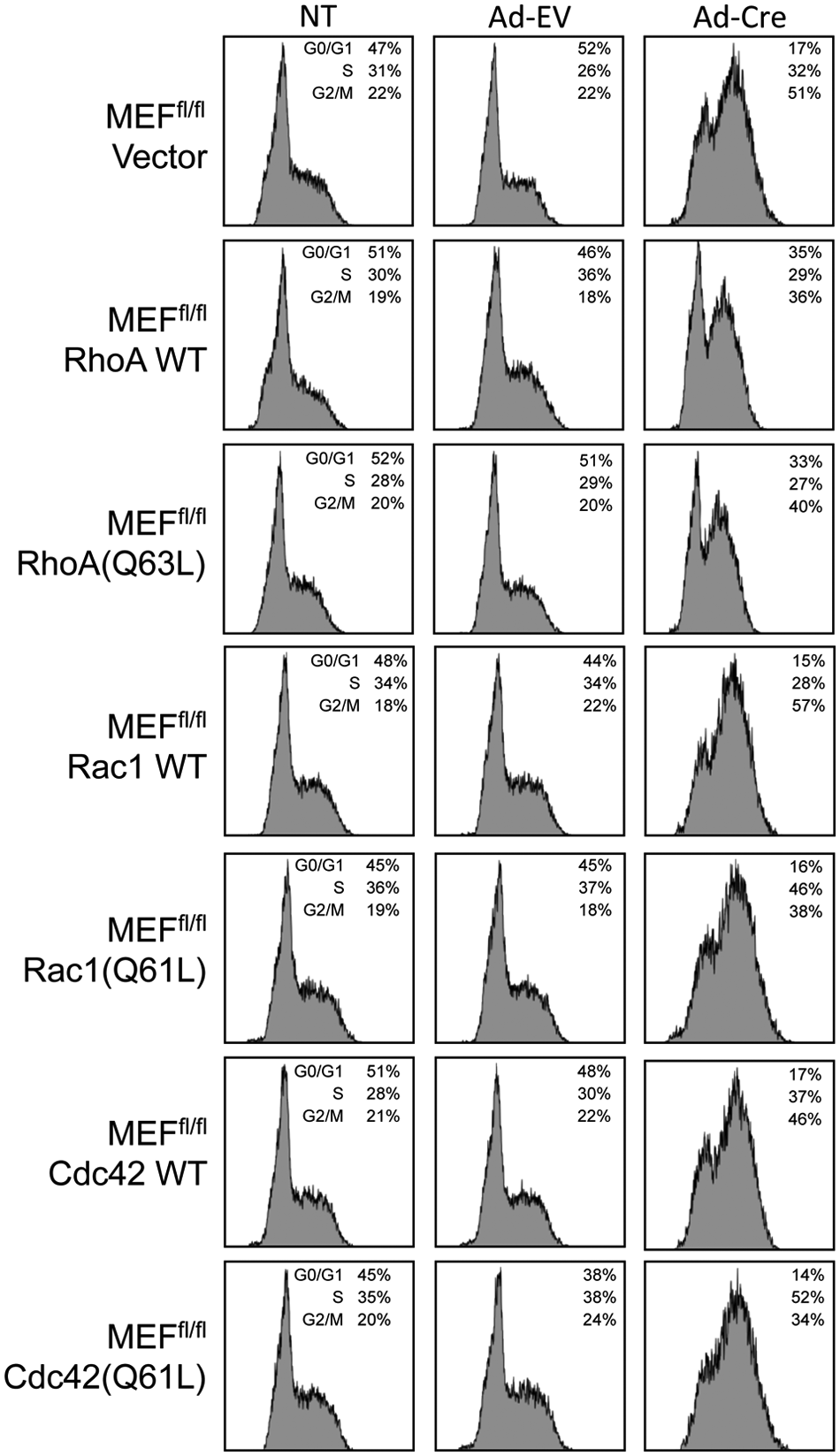
Rho GTPases did not rescue the defect cytokinesis caused by loss of Ect2. (
Discussion
Despite the fact that Ect2 is one of at least 25 RhoGEFs that activate RhoA, observations made in cell culture studies and in Drosophila 26 and Caenorhabditis elegans 27 suggest an essential and unique role for this RhoGEF in cytokinesis and cell growth. To evaluate a function for Ect2 in normal mammalian cell physiology, we generated knockout mice. We found that an Ect2 deficiency caused early embryonic lethality and a complete loss of proliferation in vitro that was associated with impaired cytokinesis, altered cell morphology, and suppressed cell migration. We conclude that Ect2 function is unique among all 83 mammalian RhoGEFs in its essential requirement for cytokinesis.
Our studies provide the first analyses of mammalian Ect2 function in normal cells in vitro and in vivo. Previously, Ect2 function in vivo was evaluated in Drosophila and C. elegans invertebrate species. In Drosophila, loss of function of the Ect2 ortholog, pebble, resulted in embryonic lethality. 26 Homozygous pebble-deficient mutant embryos at the end of embryogenesis contained fewer and larger cells with enlarged nuclei. The first 13 rapid syncytial nuclear divisions proceeded normally in pebble-deficient embryos. Following cellularization, the postblastoderm nuclear divisions occurred (mitoses 14, 15, and 16), but cytokinesis was not observed, with multinucleated cells seen at cycle 15 mitoses. In C. elegans, RNA interference of Ect2 caused sterility and embryonic lethality in addition to reduced migration of embryonic P-cells. 27 However, there is evidence for a second mammalian Dbl family RhoGEF, GEF-H1, in regulation of cytokinesis, 28 and no GEF-H1 orthologs are found in Drosophila or C. elegans. Additionally, Ect2-independent cytokinesis has been described in some human cell types. 29 Therefore, we anticipated the possibility that Ect2 may not be required for normal cell cytokinesis in vivo. However, our finding of embryonic lethality secondary to Ect2 deficiency is consistent with the essential role for this RhoGEF in normal cell cytokinesis. As Ect2 activates Rho GTPases that can be activated by other RhoGEFs, Ect2 must regulate Rho GTPases in a highly unique spatial and/or temporal pattern of activation that cannot be facilitated accurately by any other RhoGEF.
Similar to our observations with an Ect2 deficiency, a MgcRacGAP deficiency also results in preimplantation lethality. 30 MgcRacGAP is a component of central spindlin and facilitates Ect2 recruitment to the central spindle. siRNA depletion of MgcRacGAP phenocopies Ect2 loss and results in a cytokinesis defect in vitro.31-34 At E3.0 to E3.5, the MgcRacGAP deficiency caused formation of binucleated blastomeres, suggesting that MgcRacGAP is required for normal mitosis and cytokinesis in the preimplantation embryo. All homozygous mutant blastocysts failed to grow out in vitro. The similar developmental consequences of Ect2 or MgcRacGAP loss suggest that the Ect2 deficiency-induced embryonic lethality is a consequence of the role of Ect2 in cytokinesis.
A recent study addressed a role for Ect2 in mouse oocyte meiosis in vitro and found that Ect2 depletion caused a block in the extrusion of the first polar body. 35 This observation suggests a much earlier requirement for Ect2 in development than suggested from our studies. This is likely due to the presence of maternal Ect2 in the oocytes derived from Ect2 – /+ intercrosses. The requirement for Ect2 in meiosis was found to be associated with RhoA and RhoA activation of its effector, the ROCK serine/threonine kinase.
In a majority of previous studies addressing the consequence of a specific RhoGEF deficiency for normal mouse development, while specific cellular defects have been identified, generally normal development and viability were retained. 4 These observations are consistent with the redundant functions of RhoGEFs, with 83 mammalian RhoGEFs regulating 14 Rho GTPases (6 Rho GTPases are constitutively active and not believed to be regulated by RhoGEFs). 2 While 2 Dbl RhoGEFs have been found to be essential for mouse development, because they possess additional non-RhoGEF catalytic functions, it is not clear that this is due to the loss of their RhoGEF activities. For example, a Sos1 deficiency leads to embryonic lethality,36,37 but this is most likely due to the fact that Sos1 is also a RasGEF and key regulator of Ras activation by receptor tyrosine kinases. Deficiency in another Dbl RhoGEF, Trio, also causes embryonic lethality, with Trio −/− embryos dying between E15.5 and birth or shortly thereafter. 38 However, Trio is a multifunctional protein possessing 2 distinct DH-PH tandem domains, one active on RhoA and the second on Rac1, and additionally a protein serine/threonine kinase domain. Thus, whether the essential function of Trio in development is its 2 RhoGEF functions is not clear. In contrast to Sos1 and Trio, Ect2 has no other known catalytic function aside from its RhoGEF activity, although Ect2-interacting proteins may facilitate Ect2-dependent, non–RhoGEF-associated functions important for cytokinesis.
The role of Ect2 in cytokinesis has been attributed to recruitment of RhoA and localized RhoA activation at the central spindle complex.31,39 Additionally, our analyses of the isolated DH-PH domain fragment of Ect2 identified activity for RhoA, the related RhoB and RhoC isoforms, but not for 8 other Rho GTPases tested in vitro, including Rac1, Rac2, and Rac3 (data not shown). However, one study found that Ect2 was required for Cdc42 activation during mitosis. 19 Furthermore, we and others found that constitutively activated N-terminally truncated Ect2 can activate RhoA, Rac1, and Cdc42 in vivo. 40 To determine the key Rho GTPase important for Ect2 regulation of cytokinesis, we determined if ectopic expression of constitutively activated RhoA, Rac1, or Cdc42 could rescue the loss of endogenous Ect2 and restore proper cytokinesis. This approach was utilized recently to show that constitutively activated Rac1 alone could restore Ect2-dependent lung tumor cell growth. 23 Therefore, we were surprised that no one activated Rho GTPase was able to rescue the block in cytokinesis caused by Ect2 depletion. One possible explanation for this is that Ect2 may need to activate multiple Rho GTPases concurrently. Interestingly, consistent with our findings, activated RhoA orthologs did not rescue the Ect2 deficiency-induced cytokinesis defect in Drosophila 14 or C. elegans. 27 Another possible explanation may be that Ect2 activation of RhoA during cytokinesis is highly regulated spatially and temporally. Therefore constitutively activated RhoA may not accurately mimic this more precise regulation. Our ongoing evaluation of recombinant Ect2 Rho GTPase specificity found activity for only RhoA and related isoforms. Therefore, we favor the second possibility, and our future studies will address the importance of Ect2 subcellular localization in supporting cytokinesis.
In addition to a cytokinesis defect, we also observed a defect in MEF cell morphology and migration. Ect2 loss in MEFs resulted in enlarged and flattened cells with increased cytoplasmic content. Whereas wild-type MEFs are highly motile, Ect2 loss resulted in a near complete loss of movement. Similar Ect2 deficiency-associated defects were seen in normal cell migration in C. elegans and Drosophila27,41,42 and in tumor cell migration and invasion.22,23 However, these defects were associated with Rac and not RhoA activation. We observed that the Ect2 depletion-associated loss of MEF cell motility was associated with decreased membrane ruffling activity, an activity also associated with Rac activity.
In summary, our studies provide the first analysis of Ect2 function in normal cells in vitro and in vivo, and we show that, despite the existence of multiple RhoGEFs, Ect2 plays a unique role in development and progression through mitosis. Using our conditional mouse model, our ongoing studies will assess the consequences of Ect2 deficiency in adult tissue homeostasis and in tumor progression and maintenance. Furthermore, using MEFs with conditional Ect2 loss, where we can rescue the cytokinesis defect by ectopic wild-type Ect2 expression, we will perform detailed structure-function analyses of Ect2 to delineate what aspects of Ect2 are essential for its unique role in maintenance of normal cell cytokinesis.
Materials and Methods
Vector construction
The targeting vector was based on a 5.3-kb genomic fragment from the ect2 gene encompassing exons 8 to 12 and surrounding sequences. This fragment, obtained from the C57Bl/6J RP23 BAC library, was modified by inserting the distal loxP site into intron 8 and the proximal loxP site including an FRT-flanked neomycin resistance gene into intron 7. A thymidine kinase (TK) cassette was inserted at the 3′ end of the genomic fragment.
Embryonic stem (ES) cell culture
The quality-tested C57BL/6NTac ES cell line was grown on a mitotically inactivated feeder layer comprised of MEFs in Dulbecco’s modified eagle (DMEM) high-glucose medium supplemented with 20% fetal bovine serum (FBS, PAN Biotech GmbH, Aidenbach, Germany) and 1,200 U/mL leukemia inhibitory factor (ESG 1107, Millipore, Billerica, MA). One × 107 ES cells and 30 µg of linearized DNA targeting vector were electroporated (Gene Pulser, Bio-Rad Laboratories, Hercules, CA) at 240 V and 500 µF. Positive selection with G418 (200 µg/mL) started on day 2, and counterselection with gancyclovir (2 µM) started on day 5 after electroporation. Resistant ES cell colonies with a distinct morphology were isolated on day 8 after transfection and expanded in 96-well plates. Correctly recombined ES cell clones were identified by Southern blot analysis using external and internal probes and were frozen in liquid nitrogen.
Generation of mice
The animal study protocol was approved according to the German Animal Welfare Act (§8(1) TierSchG) by the local authority. Mice were kept in the animal facility at TaconicArtemis GmbH in microisolator cages (Techniplast Sealsave, Cologne, Germany). Feed and water were available ad libitum. Light cycles were on a 12:12-hour light:dark cycle with the light phasing starting at 06:00 hours. Temperature and relative humidity were maintained between 21°C to 23°C and 45% to 65%, respectively.
After administration of hormones, superovulated BALB/c females were mated with BALB/c males. Blastocysts were isolated from the uterus at dpc 3.5. For microinjection, blastocysts were placed in a drop of DMEM supplemented with 15% FBS under mineral oil. A flat-tip, piezo-actuated microinjection pipette with an internal diameter of 12 to 15 µm was used to inject 10 to 15 targeted C57BL/6NTac ES cells into each blastocyst. After recovery, 8 injected blastocysts were transferred to each uterine horn of 2.5 days postcoitum, pseudopregnant NMRI females. Chimerism was measured in chimeras (G0) by coat color contribution of ES cells to the BALB/c host (black/white). Highly chimeric mice were bred to 1) C57BL/6-Tg(CAG-Flpe)2Arte females mutant for the presence of the Flp recombinase gene or to 2) C57BL/6-Gt(ROSA)26Sor tm16(Cre)Arte females mutant for the presence of the Cre recombinase gene. This allowed detection of germline transmission by the presence of black, strain C57BL/6, offspring (G1) and creation of 1) selection marker–deleted conditional (floxed) knockout mice or 2) constitutive knockout mice by Flp- or Cre-mediated removal, in one breeding step. Chimeras bred to 3) C57BL/6 wild-type mice resulted in germline offspring with selection marker in the Ect2 genomic locus described as targeted mice.
Genotyping of mice by PCR
Genomic DNA was extracted from 1- to 2-mm-long tail tips using the NucleoSpin Tissue kit (Macherey-Nagel, Düren, Germany). Genomic DNA (2 μL) was analyzed by PCR protocol 1 in a final volume of 50 μL in the presence of 2.0 mM MgCl2, 200 μM dNTPs, 100 nM of each primer, and 2 U Taq DNA polymerase (Invitrogen, Carlsbad, CA) with the following primers: 1 (5′-GC ACTCCAATTATGAAGCCAGAATGG-3′), 2 (5′-CAATATGTTGGGTAGAGAGATGGC-3′), and 3 (5′-TCCTCCGGGTGGACCAGAG-3′) detecting the presence of the wild-type allele (335 bp), targeted allele (413 bp), conditional allele (413 bp), and constitutive allele (498 bp). Following a denaturing step at 95°C for 5 minutes, 35 cycles of PCR were performed, each consisting of a denaturing step at 95°C for 30 seconds, followed by an annealing phase at 60°C for 30 seconds, and an elongation step at 72°C for 1 minute. PCR was finished by a 10-minute extension step at 72°C. Amplified products were analyzed using 2% standard TAE agarose gels. A second PCR protocol for genotyping the Ect fl/fl mice used 2 primers: 3 (5′-TCCTCCGGGTGGACCAGAG-3′) and 4 (5′-CTGGCTTCATAATTGGAGTGC-3′). A 420-bp wild-type fragment or a 520-bp mutant fragment was amplified with these 2 primers.
For genotyping the blastocysts, a nested PCR protocol was used. The outgrowths were scraped from the well with a micropipette tip and transferred to a PCR tube. The tubes were spun down, and the supernatant was removed. The cells were resuspended in 5 μL of 400 ng/μL Proteinase K/17 μM SDS and overlayed with mineral oil. The tubes were incubated for 1 hour at 50°C and then denatured at 99°C for 30 minutes. For the first PCR reaction, 45 μL of a PCR mix containing 25 pmol of external primers (5′-CCCTCCAGGTTGAGAACTGCTACTAAG-3′ and 5′-GCAGGCTGAGAGCAAGCCAGGAGA-3′). After amplification, 1 μL of this first PCR reaction was used for a second round of PCR reactions, which used PCR protocol 1 as described above.
Blastocyst outgrowth analyses
Timed breedings of ect2 +/− breeding pairs were set up, and at E3.5, blastocysts were flushed from the uterine tract from the females. Blastocysts were placed in 20 μL of ES cell medium (DMEM, 15% FCS, 0.01% β-mercaptoethanol) in Nunc dish microwells (Nalgene, Palo Alto, CA) and grown under mineral oil in a 5% CO2 incubator at 37°C. After 7 days, the cultures were evaluated and scored for degree of outgrowth, photographed, and then genotyped by PCR.
Mouse embryonic fibroblast cultures
Ect2 fl/fl mice were crossed, and at E15.5, embryos were isolated from the female. Embryos were rinsed with phosphate-buffered saline (PBS), and the head and dark red organs were removed from the culture. The tissue was minced and resuspended in a solution of PBS/trypsin/EDTA and incubated with gentle shaking at 37°C for 30 minutes. The cell/tissue solution was gently centrifuged, the supernatant was removed, and the loose pellet was resuspended in DMEM supplemented with 10% FCS. This solution was then filtered through a cell filter and then plated onto 150-mm tissue culture plates and grown in a 5% CO2 incubator at 37°C. After 18 hours, the medium was changed to fresh growth medium. MEFs were immortalized by trypsinizing the cells every 3 days at replating on a 100-mm tissue culture plate at a density of 1 × 103 cells/dish. For knocking out Ect2 expression in the Ect fl/fl MEFs, cells were infected with recombinant adenovirus expressing Cre recombinase (Ad-Cre) with a green fluorescent protein expressing control adenovirus (Ad-GFP) used to assess Cre-specific activities (Gene Transfer Vector Core, University of Iowa, Iowa City, IA).
Constructs
An expression vector for human Ect2 was generated using a human Ect2 cDNA sequence from an expression vector kindly provided by Dr. A.P. Fields (Mayo Clinic, Rochester, MN), which was subcloned into the FUGW lentiviral expression vector, 43 a gift from Dr. Bryan Roth (University of North Carolina at Chapel Hill, Chapel Hill, NC). Mammalian expression vectors of wild-type human Rho GTPases (RhoA, Rac1, and Cdc42) were constructed by ligation of cDNA sequences into a pBabe-puro retrovirus expression vector 44 that introduces an N-terminal HA epitope tag. The constitutively active (RhoA(Q63L), Rac1(Q61L), and Cdc42(Q61L)) and fast-cycling Rho GTPase mutants (RhoA(F30L), Rac1(F28L), and Cdc42(F28L)) were created using QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA).
Western blot
To evaluate protein expression, 4 days after Cre infection, the MEFs were lysed and analyzed by SDS-polyacrylamide gel electrophoresis and Western blot analysis, with anti-Ect2 antibody (C-20, Santa Cruz Biotechnology, Santa Cruz, CA), anti-HA (Covance, Princeton, NJ), and anti–β-actin (Sigma, St. Louis, MO) as a loading control. Additionally, we utilized a second Ect2 polyclonal antibody generated against KLH-conjugated linear peptide corresponding to C-terminal sequences in human Ect2 (07-1364, Millipore).
Immunofluorescence
To visualize nuclei content, 3 days after Ad-Cre infection, the MEFs were plated onto coverslips. Twenty-four hours after attachment, coverslips were rinsed with PBS and fixed with 2% paraformaldehyde in PBS for 10 minutes. The cells were stained for 5 minutes with 5 ng/mL 4′,6-diamidino-2-phenylindole (DAPI) in PBS, washed 2 to 3 times with PBS, and then mounted onto slides. The cells were visually analyzed for nuclei content on an Olympus BX61 upright fluorescence microscope (Tokyo, Japan). Images were collected using Velocity (PerkinElmer, Waltham, MA). To visualize polymerized F-actin, cells were stained with Alexa Fluor 647 phalloidin (Invitrogen).
Flow cytometry
Cells were trypsinized, washed in PBS, then fixed in 70% ethanol, and stored at −20°C. The day prior to analysis, the cells were washed in PBS and then stained with 0.5 mL of propidium iodine staining solution (1% Triton-X-100, 1 mg/mL PI, 100 mg/mL of RNAse A) overnight at 4°C. Data were collected on a Beckman Coulter CyAn analyzer (Brea, CA), and ModFit (Becton Dickinson) was used for cell cycle analysis.
Footnotes
Acknowledgements
The authors thank Alan Fields for the cDNA sequence for human Ect2 and Bryan Roth for the FUGW lentiviral expression vector.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This study was supported by National Institutes of Health grants CA129610 and CA67771 (to C.J.D.) and CA140424 (to J.J.Y.). D.R.C. was supported by a T32 CA71341 training grant and an F31 predoctoral fellowship (CA159821). G.K. and M.S. are employees at TaconicArtemis, Cologne, Germany. The Ect2 knockout model was developed at Artemis for Exelixis. The animal model was characterized by University of North Carolina at Chapel Hill, under the premise of an academic collaboration.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.