
Abstract
Emerging data suggest that SSeCKS/Gravin/AKAP12 (“AKAP12”), originally identified as an autoantigen in cases of myasthenia gravis, controls multiple biological processes through its ability to scaffold key signaling proteins such as protein kinase (PK) C and A, calmodulin, cyclins, phosphoinositides, “long” β-1,4 galactosyltransferase (GalTase) isoform, Src, as well as the actin cytoskeleton in a spatiotemporal manner. Specialized functions attributed to AKAP12 include the suppression of cancer malignancy, especially aspects of metastatic progression, regulation of blood-brain and blood-retina barrier formation, and resensitization of β2-adrenergic pain receptors. Recent data identify a direct role for AKAP12 in cytokinesis completion, further suggesting a function as a negative regulator of cell senescence. The current review will discuss the emerging knowledge base of AKAP12-related biological roles and how the factors that affect AKAP12 expression or that interact with AKAP12 at the protein level control cancer progression and blood-tissue barrier formation.
Keywords
Introduction
What was originally identified as a minor autoantigen correlating with poor prognosis in myasthenia gravis, and thus called Gravin, 6,7 was subsequently shown to be orthologous to a rodent protein, SSeCKS (pronounced essex), a major PKC substrate and binding protein whose expression is suppressed in Src- and Ras-transformed cells. 10 The demonstration that Gravin and SSeCKS also bind RII isoforms of PKA 11 led to the renaming of these orthologs to A kinase anchoring protein (AKAP)-12. Importantly, AKAP12 orthologs have been identified in all vertebrate species analyzed including Danio and Fugu, but no credible orthologs have been found in insects, fungi, or worms. Whereas mouse and rat SSeCKS are >90% identical at the protein level, rodent SSeCKS is 83% identical to human AKAP12 over the N-terminal (approximately 1,000 a.a.) and 40% identical to the Xenopus ortholog, Xgl, over the N-terminal (800 a.a.). 14 Vertebrates encode only a single copy of their AKAP12 ortholog, and whole genome sequencing has failed to identify homologous gene family members within a given species. In humans, AKAP12 maps to 6q24-25.2, a deletion hotspot in cases of advanced prostate, breast, and ovarian cancer. 14 This region is syntenic with the telomeric end of mouse chromosome 10, and indeed, mouse AKAP12 maps to this region (www.ensembl.org).
In humans and rodents, 2 major AKAP12 transcripts, α and β, are expressed from independent promoters spaced roughly 35 Kb apart; yet, they splice to a common large exon that results in their sharing >93% amino acid identity. 19 Whereas α and β AKAP12 protein isoforms are expressed ubiquitously in the embryo and the adult as 305- or 290-kDa products, respectively (290 or 280 kDa in rodents), there is high expression in some epithelial populations such as in the prostatic luminal cells, in some specialized cells such as glomerular mesangial cells and podocytes, and in Purkinje cells in the brain. The highest organ-specific expression is detected in the testes, ovary, brain, lung, prostate, and cardiac muscle. In contrast, a smaller testes-specific γAKAP12 isoform shares roughly 85% of the common C-terminal α and β residues. 20 AKAP12-null mice, deficient in all 3 major isoforms, are viable, although they suffer from delayed fertility and prostatic hyperplasia. 21 In contrast, the loss of AKAP12 causes selective gastrulation defects in Danio embryos, 22 although it is unclear whether these are sufficient to prevent development of adult fish.
AKAP12 Scaffolding Functions
Scaffolding proteins are defined by their ability to bind signaling and/or cytoskeletal proteins in a spatiotemporal manner, typically involving scaffolding protein multimerization and the ability to associate with one or more specific cellular sites. Association with plasma and vesicle membrane sites by AKAP12 is facilitated by N-terminal myristylation, found only in the α isoform, 1 and by 3 so-called polybasic effector domains 13 that likely bind phosphoinositides and other phospholipids based on their homology to a membrane-binding domain in the MARCKS protein. 23 Indeed, mouse AKAP12 was identified independently as a phosphatidylserine-binding protein, 16,24 a function that maps to the effector domains and that facilitates association with plasma membranes and PKC. 13 The 3 effector domains are required for AKAP12 to mediate cell flattening. 25
Although AKAP12 is a weak F-actin cross-linking protein in vitro, it co-stains with F-actin, and this association can be antagonized by AKAP12 tyrosine phosphorylation via an EGF- or PDGF-induced, FAK-dependent pathway. 4 Recent data suggest that AKAP12 may also associate with the actin or tubulin cytoskeleton, respectively, via direct or indirect association to profilin or dynein (below). Evidence that AKAP12 multimerizes comes from the Scatchard analysis of AKAP12/F-actin in vitro binding 4 and from protein-protein interaction databases such as MINT (http://mint.bio.uniroma2.it/mint/).
AKAP12 binding partners have been identified by homology searching, yeast 2-hybrid analysis, and mass spectrometry of coimmunoprecipitating proteins. To date, protein binding domains have been identified that bind PKC, PKA-RII, cyclins, calmodulin, β1,4-galactosyltransferase-polypeptide 1 (B4galT1), Src, β2-adrenergic receptor, 26 F-actin, and cAMP-specific 3′,5′-cyclic phosphodiesterase 4D 27 (Fig. 1). Recent large-scale protein-protein interaction screens identified associations with profilin (PFN1), pyruvate kinase M2 fragment (PKM2), premature ovarian failure protein (POF1B), peptidyl prolyl isomerase (PPIA; cyclophilin-A), C1 segment protein (SC-9C5.12), small proline-rich protein II (SPRR2E), and transferring (TF) and valosin-containing protein (VCP), although specific binding sites on AKAP12 have not yet been characterized (http://www.mitocheck.org/cgi-bin/mtc?query=MCG_0000007). Association with SC-9C5.12, which binds dynein, would facilitate interaction between AKAP12 and the tubulin cytoskeleton, whereas association with VCP, which binds Cdc42, might facilitate AKAP12 association with the leading edge of motile cells. Finally, the ability of AKAP12 to associate with and control the cytokinesis apparatus might be facilitated by its interaction with transferrin, which in turn binds a MIS12/Bub1 complex. Importantly, there is evidence that phosphorylation of AKAP12 by mitogen-induced kinases antagonizes AKAP12 scaffolding activity, whereas phosphorylation by kinases associated with differentiation, such as PKA, might enhance AKAP12 scaffolding activity. Both the serine and tyrosine phosphorylation of AKAP12 increases during G1→S progression and then decreases precipitously in G2/M, 4,28 even though there is only a small decrease in total AKAP12 protein levels during these phases. 28 Thus, prephosphorylation of AKAP12 by PKC decreases its ability to bind PKC, calmodulin, and cyclins in vitro and in cellulo. 8,15 In contrast, PKA can phosphorylate AKAP12 and enhance association with a Src SH3 domain, thereby facilitating β2-adrenergic receptor complex resensitization. 29-32 Whereas AKAP12 binding to PKC inhibits its kinase activity 1,2,11 and sequesters PKC isozymes at plasma membrane sites, AKAP12 regulates cyclin D, in part, by sequestering cyclin D pools in the cytoplasm. 8 Interestingly, SSeCKS phosphorylation by PKC in neuronal cells causes a decrease in AKAP12-PKC scaffolding but no change in AKAP12-PKA binding 33 or agonist-induced PKA activation, 11 suggesting that AKAP12 controls the well-recognized, mutually exclusive activation cross-talk between PKC and PKA. The differential PKC-PKA control likely relates to overlap between PKC binding and phosphorylation sites mapping to the N-terminus of AKAP12, in contrast to the PKA-RII binding site mapping to a C-terminal region lacking phosphorylation sites (Fig. 1). Indeed, the C-terminal AKAP site is required for AKAP12 to target PKA-RII to the cell periphery, 34 and PKC activation by phorbol esters causes the AKAP12/PKA-RII complex to translocate to the perinucleus. 13,33 Lastly, in round spermatids, AKAP12 co-stains with calcineurin. 35
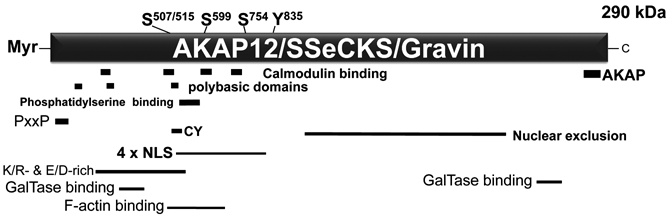
AKAP12 scaffolding domains. Known PKC phosphorylation sites 1,2 are identified at top as Ser507/515, 599, and 748 (based on the rat protein sequence: NP_476444.2) as well as an implied EGF- or PDGF-induced, FAK-dependent tyrosine phosphorylation site at Y835. 4 A Src SH3 binding site (P15xxP). 5 AKAP12 also encodes at least 4 nuclear localization signals (NLS) homologous to those from SV40 Tag, a cyclin binding domain containing 2 CY motifs, 8 an acidic/basic region (KR- and E/D-rich), an A kinase anchoring protein (AKAP) domain for PKA RII isoform binding, 11 a nuclear exclusion domain, 12 3 polybasic domains involved in membrane association, 13 at least 4 calmodulin binding domains of the so-called 1-5-10 motif, 15 a phosphatidylserine (PS) binding domain 16 that facilitates association with PKC isoforms, 17 2 distinct binding domains for β1,4-galactosyltransferase (B4GALT1), 18 and an F-actin binding domain. 4
AKAP12 protein levels increase >10-fold during contact inhibition, 8,28,36 and the relative low basal and mitogen-inducible phosphorylation of AKAP12 under this condition 4,28 suggests that its scaffolding activity for signaling proteins, and thus its activity as a negative mitogenic regulator (below), is highest during contact inhibition.
AKAP12 Functions in Normal Cell Cycle: Regulator of Mitogenesis and Cytokinesis
Several lines of evidence suggest that AKAP12 functions in normal cells to 1) prevent inappropriate cell cycle progression and 2) facilitate completion of cytokinesis and mitosis. Early attempts to constitutively express AKAP12 10 led to the finding that its overexpression in untransformed fibroblasts 8 and epithelial cells 36 caused G1 arrest due to the suppression of serum-induced, MEK/ERK-dependent cyclin D expression. 8 Interestingly, AKAP12 encodes 2 so-called cyclin binding (CY) motifs, which can also cause G1 arrest by sequestering pools of cyclin D in the cytoplasm during contact-inhibited growth. 8 PKC-induced phosphorylation of AKAP12 at sites within the CY domains antagonizes AKAP12-cyclin binding, 15 resulting in the nuclear translocation of cyclin D even in contact-inhibited cultures, followed by one additional round of DNA replication. 8 Recent data indicate that compared to WT mouse embryo fibroblasts (MEF), AKAP12-null MEF have higher basal- and serum-induced MEK/ERK activation, rates of serum-induced proliferation, saturation densities, and autocrine growth associated with increased basal cyclin D levels (Akakura S, Nochajski P, Gao L, Sotomayor P, Matsui S, Gelman IH. Rb-dependent cellular senescence, multinucleation and susceptibility to oncogenic transformation through PKC scaffolding by SSeCKS/AKAP12. Cell Cycle: in press.) (Gao and Gelman, unpublished data).
A pool of AKAP12 may help facilitate completion of cytokinesis during normal cell cycle progression. Recent data indicate that although AKAP12-null MEF initially proliferate faster than WT MEF, they suffer from premature senescence, exhibiting binucleation or polyploidy. This senescence is Rb but not p53 dependent, and it correlates with increased PKCα and δ activity due to the loss of AKAP12 scaffolding. Activated PKCδ causes the downregulation of Lats1, a mitotic exit network kinase required for completion of cytokinesis. 37 In parallel, higher PKCα kinase levels induce p16Ink4a expression and Rb activation by increasing MEK kinase activity, which in turn downregulates expression of Id1, a negative regulator of p16Ink4a expression (Akakura S, Nochajski P, Gao L, Sotomayor P, Matsui S, Gelman IH. Rb-dependent cellular senescence, multinucleation and susceptibility to oncogenic transformation through PKC scaffolding by SSeCKS/AKAP12. Cell Cycle: in press.). Indeed, higher AKAP12 protein levels are found in senescent human diploid fibroblasts and in aging rat and human keratinocytes, 38 and the siRNA-mediated knockdown of AKAP12 is sufficient to induce senescence associated with binucleation (Akakura S, Nochajski P, Gao L, Sotomayor P, Matsui S, Gelman IH. Rb-dependent cellular senescence, multinucleation and susceptibility to oncogenic transformation through PKC scaffolding by SSeCKS/AKAP12. Cell Cycle: in press.). Moreover, AKAP12 may directly regulate cytokinesis based on the demonstration of a pool staining in the anaphase abscission furrow, 39,40 and that loss of AKAP12 leads to multinucleation. 39,41 The furrow contains the actin-myosin ring whose PKC-RhoGTPase–dependent contraction helps complete daughter chromosome separation, 42 and thus, it is conceivable that AKAP12 might regulate cytokinesis via its ability to scaffold PKC and actin and to attenuate RhoGTPase activity. 43 Strengthening this notion, a recent systems biology analysis predicts the existence of a mitotic protein complex containing AKAP12, phosphodiesterase 4D, ATM, Polo-like kinase 4, APC, dynein, and profilin (complex MCC_0000069; http://www.mitocheck.org/cgi-bin/mtc?query=MCG_0000007).
AKAP12 and Cell Motility
Overexpression and knockdown experiments indicate that AKAP12 likely does not control generic cell motility but that it might attenuate specialized motility in response to specific chemoattractants. For example, AKAP12-re-expressing MAT-LyLu prostate cancer cells have normal short- and long-term motility in monolayer wound-healing assays. 36 In contrast, AKAP12 inhibits chemotaxis in these same cells due to the inhibition of a PKC-Raf/MEK/ERK pathway, 44 and similarly, AKAP12-null MEF display higher chemotaxis towards serum or PDGF-BB than passage-matched WT MEF (Hyun-Kyung Ko and Irwin Gelman, unpublished data). A recent study by Busch et al. 45 strongly suggests that upregulated AKAP12 facilitates HGF-induced, c-Met–dependent cell motility through the upregulation of PKA activity and PKA-induced genes, presumably through AKAP12’s scaffolding function. Taken together, these data strengthen the notion that AKAP12 facilitates the differential activation of PKC and PKA in processes such as cell motility.
Consistent with AKAP12 being a cytoskeletal protein that can regulate the remodeling of the actin cytoskeleton, 46 either AKAP12 overexpression or deficiency in untransformed cells causes cell flattening 3,8,9,46 (Fig. 2). However, whereas overexpression antagonizes stress fiber formation and leads to the production of long filamentous projections, AKAP12 deficiency results in the production of thickened, longitudinal stress fibers and increased numbers of focal adhesion plaques. Indeed, in the absence of FAK, AKAP12 co-stains with stress fibers, whereas in EGF- or PDGF-stimulated FAK+/+ cells, AKAP12 stains at the plasma membrane and along a cortical cytoskeleton. 4 It is reasonable to speculate that the dynamic exchange of AKAP12 between filamentous and cortical cytoskeletal networks plays a role in controlling specialized cell motility. Taken with the ability of AKAP12 to normalize actin cytoskeletal structures in cancer cells (below), these data suggest that AKAP12 functions as a morphostat, that is, a protein that regulates oncogenic and specialized motility pathways through the normalization of cytoskeletal architecture.
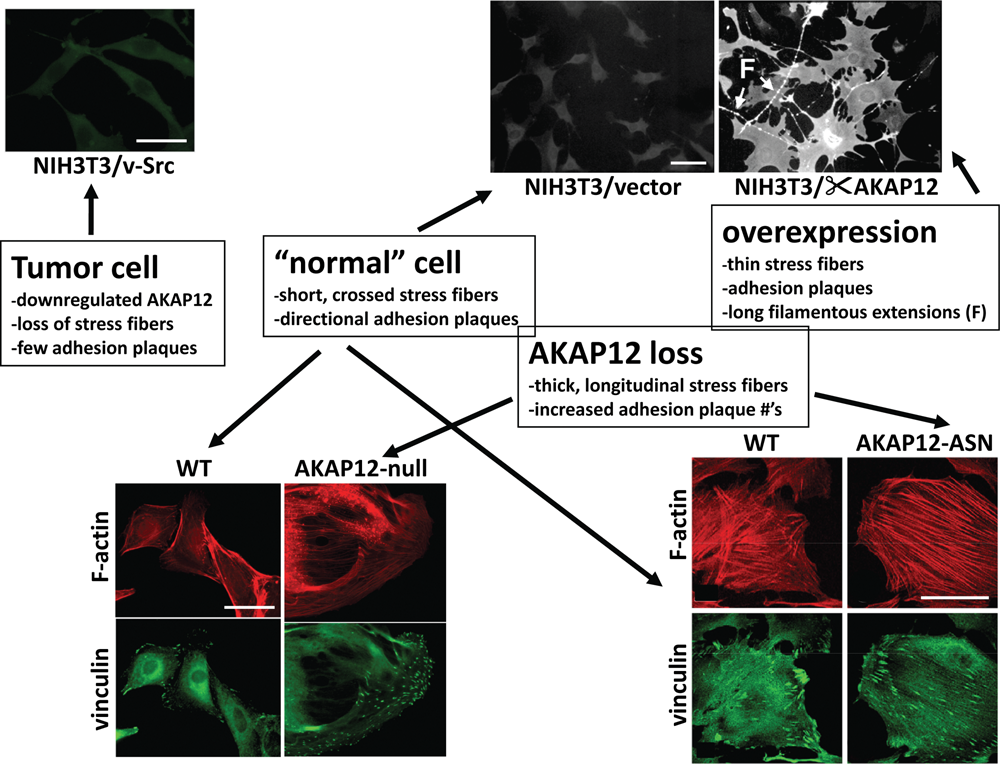
Control of cell shape and cytoskeletal dynamics by AKAP12. The downregulation of AKAP12 in v-Src–transformed NIH3T3 murine fibroblasts correlates with a transition from the polygonal morphology of parental NIH3T3 (top panel, middle) to a so-called fusiform morphology (top panel, left). In contrast, overexpression of AKAP12 via a Tet-OFF vector system 3 causes cell flattening, the thinning of stress fibers, and the production of long filamentous (F) projections (top panel, right). The staining intensities in the top panel reflect relative levels of AKAP12. AKAP12 loss in MEF (bottom left) or in stellate mesangial cells due to treatment with antisense (ASN) AKAP12 oligonucleotides (bottom right) results in cell flattening marked by thickened, longitudinal stress fibers (“F-actin”) and increased numbers of focal adhesion plaques (“vinculin”). 9
AKAP12 as a Tumor/Metastasis Suppressor
Evidence that AKAP12 might be a tumor and/or metastasis suppressor comes from its mapping to 6q24-25.2, a known deletion hotspot in many cancers, 36 its downregulation by specific oncogenes and in cancer cell lines and human cancer tissues compared to normal controls, its upregulation by treatments that suppress oncogenic growth, by direct demonstrations showing that re-expression suppresses in vitro and in vivo oncogenic growth, especially metastasis formation, or that its loss produces a tumor- or metastasis-prone condition.
An early identification of AKAP12 was based on its severe downregulation in Src-transformed fibroblasts 1 and the finding that its re-expression could suppress in vitro parameters of oncogenic growth. 3,36 Later studies showed 5- to 15-fold AKAP12 downregulation in fibroblasts and epithelial cells transformed by oncogenic versions of Ras, 1,47 Myc, 1,48-50 Jun, 51 Fos and Dnmt1, 47 and Wnt I, 52 but not by oncogenic Raf, Mos, or Neu. 10 This indicates that AKAP12 loss is not a generic effect of oncogenic transformation but due to specific oncogenic pathways. Moreover, AKAP12 levels track with transformation status; namely, they are downregulated following Ras transformation, they rise to normal levels in revertants, and then they are downregulated in retransformants, correlating especially with anchorage-independent growth. 3 Whereas WT MEF require two or more oncogenes for efficient transformation, 53 AKAP12-null MEF can be transformed efficiently by single oncogenes such as v-Src or Ras, strengthening the idea that AKAP12 functions in cell culture systems as a tumor suppressor gene (Akakura S, Nochajski P, Gao L, Sotomayor P, Matsui S, Gelman IH. Rb-dependent cellular senescence, multinucleation and susceptibility to oncogenic transformation through PKC scaffolding by SSeCKS/AKAP12. Cell Cycle: in press.). A recent study showed that downregulation of the αAKAP12 promoter by Src requires a short proximal sequence that binds USF1, Sp1, Sp3, HDAC1, 54 plus a form of TFII-I that is tyrosine phosphorylated by activated Src. 55
AKAP12 expression is also induced by genes or treatments that antagonize oncogenic transformation. For example, AKAP12 is upregulated by re-expression of the p53 tumor suppressor, 56 Smad-4–dependent, TGF-β–induced cell cycle arrest, 57-59 by antagonizing STAT3β-dependent oncogenic signaling, 60 and by differentiating agents that suppress tumor growth such as vitamin D3 analogs 61,62 and retinoids 63,64 (Sunamoto and Gelman, unpublished observations; Streb JW, Long X, Lee T-H, Sun Q, Kitchen CM, Georger MA, Metlay LA, Blaner WS, Carr DW, Gelman IH, Miano JM. Retinoid-induced expression of an immediate early tumor suppressor gene in vascular smooth muscle cells in vitro and in vivo. Submitted for publication.). C/EBPα-induced growth arrest in BCR-ABL–positive KCL22 cells results in a 15-fold increase in AKAP12 expression. 65 AKAP12 expression is induced by androgen levels that cause growth arrest in untransformed human prostate epithelial cells and in androgen-dependent LNCaP cells, 66,67 whereas selection of androgen-independent LNCaP cells correlates with AKAP12 downregulation. 68 AKAP12 expression can be stimulated by the forced expression of the GATA-3 transcription factor, which normally facilitates breast development but which is downregulated during progression to malignancy. 69
Re-expression of AKAP12 suppresses many parameters of oncogenic growth in cell culture assays. For example, AKAP12 re-expression suppressed v-Src-, Jun- or Ras-induced anchorage- and growth factor–independent growth, focus formation, Matrigel invasiveness, and podosome formation, while re-establishing normalized cytoskeletal structures such as stress fibers and mature focal adhesion plaques. 28,36,43,51 AKAP12 inhibits neither Src autophosphorylation activity during this suppression 3,43 nor Src’s ability to phosphorylate signaling proteins such as Shc or invasion-related proteins such as Tks5. 43,44 However, AKAP12 does inhibit invasiveness and chemotaxis through disengaging Src from activating downstream PKC-Raf/MEK/ERK pathways controlling cytoskeletal remodeling, podosome formation, and MMP-2/9 expression. 44 Choi et al. 70 were also able to reverse several in vitro oncogenic growth parameters of AGS gastric cancer cells by the re-expression of human AKAP12.
AKAP12 can be defined as a metastasis suppressor based on the currently accepted definition of a gene that fails to significantly affect primary tumor growth but which suppresses one or more parameters of metastasis. 71 Thus, the tetracycline-regulated re-expression of AKAP12 in MAT-LyLu rat prostate cancer cells causes a small decrease in primary subcutaneous tumors yet severely suppresses the formation of macroscopic lung metastases. 36 Interestingly, AKAP12 re-expression did not alter lung colonization activity, but it did result in relatively avascular micrometastases. 72 The finding that astrocyte-encoded AKAP12 can suppress JNK-dependent VEGF expression and angiogenesis during the establishment of the blood-brain barrier right after birth 57 led to our follow-up study, which showed that AKAP12 could suppress the expression of VEGF and other proangiogenic genes in tumor and mural cells and that the forced expression of VEGF-A (165 or 121 isoforms) in the AKAP12-re-expressing MAT-LyLu cells could partially rescue formation of lung macrometastases. 72 AKAP12’s role as a metastasis suppressor is strengthened by multiple gene expression array studies (www.oncomine.org) showing decreased levels of AKAP12 transcripts in metastases versus primary-site colon, endocrine, and breast cancers (Fig. 3) or versus primary prostate cancers (Ray M, Zheng S, Nochajski P, Davis W, Mohler JL, Marshall J, Gelman IH. Loss of SSeCKS/Gravin/AKAP12 expression in prostate cancer tissues correlates with disease progression. Submitted for publication.) or in predicting metastasis of bladder cancer after 1 year (Fig. 3).
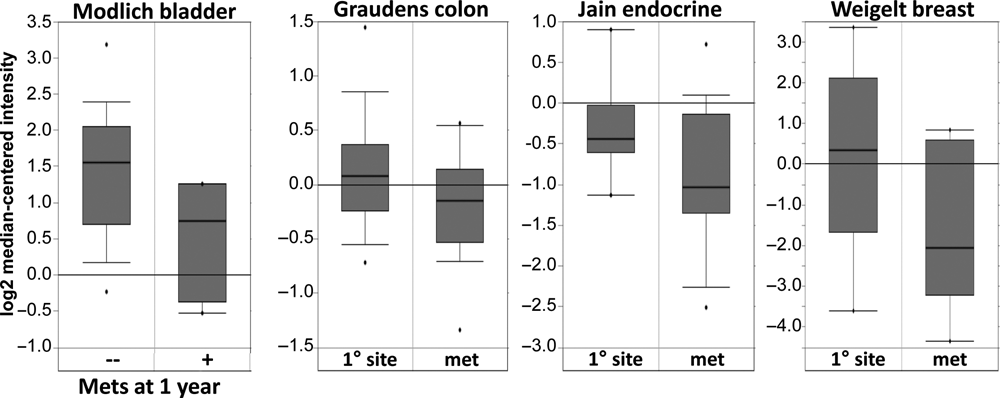
Decreased AKAP12 expression in metastatic progression. Oncomine study data showing statistically lower AKAP12 RNA expression levels in metastases compared to primary-site colon, endocrine, or breast cancers, as well as lower AKAP12 RNA levels correlating with incidence of metastasis formation at 1 year after primary diagnosis (left).
Downregulation of AKAP12 in Human Cancers
A large corpus of data indicates that AKAP12 expression is downregulated in many solid and liquid cancers types, either associated with gene deletion or epigenetic downregulation due to promoter hypermethylation or changes in chromatinization. For example, AKAP12 staining is consistently lost in prostate cancers with Gleason sum >6 36 (Ray M, Zheng S, Nochajski P, Davis W, Mohler JL, Marshall J, Gelman IH. Loss of SSeCKS/Gravin/AKAP12 expression in prostate cancer tissues correlates with disease progression. Submitted for publication.).
Roughly 40% of the Gleason sum 7 to 10 lesions that were deficient AKAP12 staining also exhibited gene deletion, as determined by laser capture microdissection followed by PCR analysis (AKAP12 and GAPDH control primers) (Gelman, unpublished data). Reports showing cancer-related loss of AKAP12 expression include human and rat prostate cancer cell lines, 36 pulmonary adenocarcinomas, 73 leiomymoma, 74 chronic and acute myeloid leukemias and myelodysplastic syndromes, 75-77 multiple myelomas, 78 papillary thyroid carcinoma, 79 pediatric acute lymphoblastic leukemia, 80 gastric cancer, 70 non-small cell lung carcinoma, 81 osteosarcomas, 82,83 melanomas, 84,85 retinoblastomas, 86 colon cancer, 87,88 fibrosarcomas, 89 and squamous cell lung carcinoma. 90,91 In all these cases, AKAP12 transcript levels are suppressed 5- to 15-fold compared to matched controls. Many microarray-based studies demonstrate 3- to 10-fold reduction in relative AKAP12 mRNA levels in breast, prostate, lung, and ovarian cancers and in gliomas, 77,92-100 and others have been cited in Entrez GEO (Gene Expression Omnibus; www.ncbi.nlm.nih.gov/geo) or Oncomine (www.oncomine.org) linking AKAP12 expression with tumor suppression. In silico analyses at the Cancer Genome Anatomy Project SAGE (Serial Analysis of Gene Expression) site (http://cgap.nci.nih.gov/SAGE) identify AKAP12 downregulation in human thyroid, lung, and liver cancer tissue and in human ovarian cancer cell lines. The majority of cancers suffering from epigenetic AKAP12 silencing seem to be regulated via promoter hypermethylation, 70,78,85,88,99,100 although instances exist where AKAP12 silencing is through changes in histone acetylation. 78 Indeed, the silencing of AKAP12 via promoter hypermethylation 101,102 has been shown to function as a neoplastic progression biomarker in Barrett’s esophagus. 103
In contrast to these examples of AKAP12 downregulation in cancer, AKAP12 expression is moderately induced in several cancer cell lines and human cancers, likely reflecting the varying biologies and/or activated signaling pathways of these cancer types. These include chronic myelogenous leukemia, 104 the pancreatic cancer cell lines PANC-1 and SU8686 versus normal mucosa, 105 muscle-invasive versus superficial bladder cancer, 106 giant cell granuloma of the jaw, 107 and high-grade follicular lymphoma. 108
Control of Neovascularization and Barriergenesis
AKAP12 can attenuate neovascularization and barrier formation, such as the blood-brain (BBB) or blood-retina (BRB) barriers, through the downregulation of proangiogenic genes such as those encoding HIF-1α or VEGF. The first such study identified AKAP12 upregulation during normoxic transition of the mouse embryo right at birth and showed that AKAP12 was responsible for suppressing angiogenesis through a JNK-dependent downregulation of VEGF and for inducing formation of the BBB after birth. 57 Indeed, chemotherapies that paradoxically induce angiogenesis result in increased VEGF and decreased AKAP12 mRNA levels, 109 strengthening the idea that AKAP12 is an antagonist of VEGF. Our data showing that AKAP12 re-expression leads to the suppression of neovascularization at metastatic sites through the downregulation of VEGF 72 further strengthens this concept. AKAP12 seems to strengthen BRB formation by upregulating tight junction proteins and by decreasing VEGF expression through the enhancement of HIF-1α/VHL binding. 86 Interestingly, the studies above show that AKAP12 can suppress BBB, BRB, and metastasis-related angiogenesis both when expressed ectopically in barrier-forming endothelial cells or through the induction of secreted factors after AKAP12 expression in astrocytes or stromal cells. Moreover, the ability of AKAP12 to secrete antiangiogenic and probarriergenic factors requires the suppression of PKC and RhoGTPase/Rho kinase activity, 110 presumably through a direct scaffolding of PKC by AKAP12. Consistent with the notion that PKC-mediated phosphorylation of AKAP12 antagonizes its scaffolding activity, You et al. showed that the ability of IL-17F, IL-1β, or TNFα to induce permeability of microvascular endothelial layers correlated with the activation of PKC and the hyperphosphorylation/downregulation of AKAP12. 111,112
Conclusion
AKAP12 likely regulates cell cycle progression, specialized cell motility, and angiogenesis by controlling actin cytoskeletal remodeling and signaling pathways through its multiple scaffolding domains (Fig. 4). (Similar functions are thought to be involved with AKAP12 regulation of β2-adrenergic receptor resensitization. 32,113 ) Current data indicate that AKAP12 can regulate Raf/MEK/ERK pathways through direct scaffolding functioning upstream of ERK, JNK, and PKC signaling. Suppression of oncogenic proliferation, invasiveness, chemotaxis, neovascularization, and cell senescence all likely involve attenuation of PKC activation through AKAP12’s spatiotemporal scaffolding functions. Some of these may also involve the downstream attenuation of Raf/MEK/ERK signaling, such as in specialized motility or MMP, HIF-1α or VEGF expression. Other AKAP12-regulated parameters, such as podosome formation, are downstream of PKC and RhoGTPases but independent of MEK, and yet, others involve PKC- and MEK-independent JNK pathways, such as VEGF and MMP secretion.
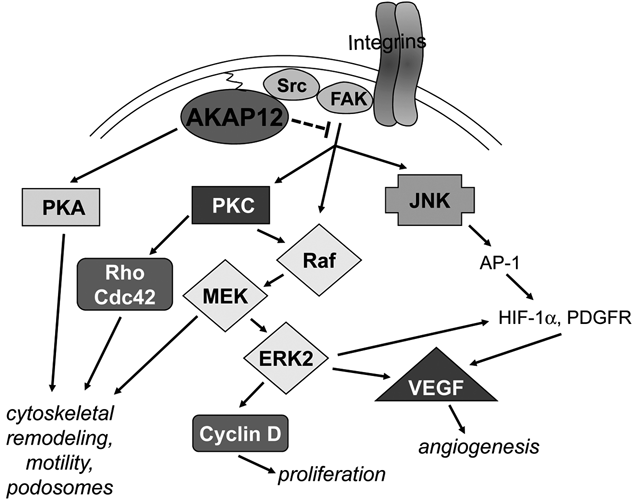
Pathways controlled by AKAP12 in cancer. AKAP12 suppresses cancer progression by disengaging adhesion- and growth factor–induced activation of Src-FAK complexes from transducing Raf/MEK/ERK- and JNK-mediated proliferation, angiogenesis, and cytoskeletal remodeling signals, possibly through a direct binding of AKAP12 to the Src-SH3 domain, resulting in the altering of Src-FAK signaling complexes. AKAP12 also directly inhibits PKC activation by a direct scaffolding function. In contrast, AKAP12 scaffolding of PKA facilitates cAMP-induced cytoskeletal remodeling.
How these pathways are regulated by AKAP12 in the context of metastasis remains unclear. However, the growing evidence implicating AKAP12 in the regulation of specialized biologies such as chemotaxis, invasiveness, and neovascularization at metastatic sites, possibly through antagonizing required Src-signaling pathways, 114,115 strongly suggests that AKAP12 regulatory functions are heavily influenced by specific microenvironmental conditions. Thus, although AKAP12 expression is high in many tissues, the loss of its expression in transgenic mice results in hyperplasia and/or dysplasia in only some sites. 21 Similarly, this may be why AKAP12 downregulation plays a direct role in metastasis suppression in specific tumor sites such as the prostate or pancreas. 116
Footnotes
Acknowledgements
The author thanks Andrei Bakin for critical reading of this article. This paper is dedicated to the memory of my postdoctoral mentor, Dr. Hidesaburo Hanafusa, who taught me that scientific advancement required a keen sense of observation, faithfulness to detail, dogged perseverance, and the humility to withstand strong critical review from the outside as well as even stronger criticism from within.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by the National institutes of Health [grant number CA94108]; Department of Defense [grant numbers PC074228, PC061246, BC086529]; and the Roswell Park Alliance Foundation.