
Abstract
We tallied the number of possible mutant amino acids in proteins thought to be inactivated early in tumorigenesis and in proteins thought to be inactivated late in tumorigenesis, respectively. Proteins thought to be inactivated early in tumorigenesis, on average, have a greater number of alternative, mutant possibilities, which raises the possibility that the sequential order of mutations associated with cancer development reflects the random chance, throughout life, of a mutagen inactivating a larger versus a smaller target. The hypothesis that the temporal order of genetic changes in cancer reflects mutagen target sizes leads to novel considerations of 1) the mechanisms of the acquisition of cancer hallmarks and 2) cancer screening strategies.
Introduction
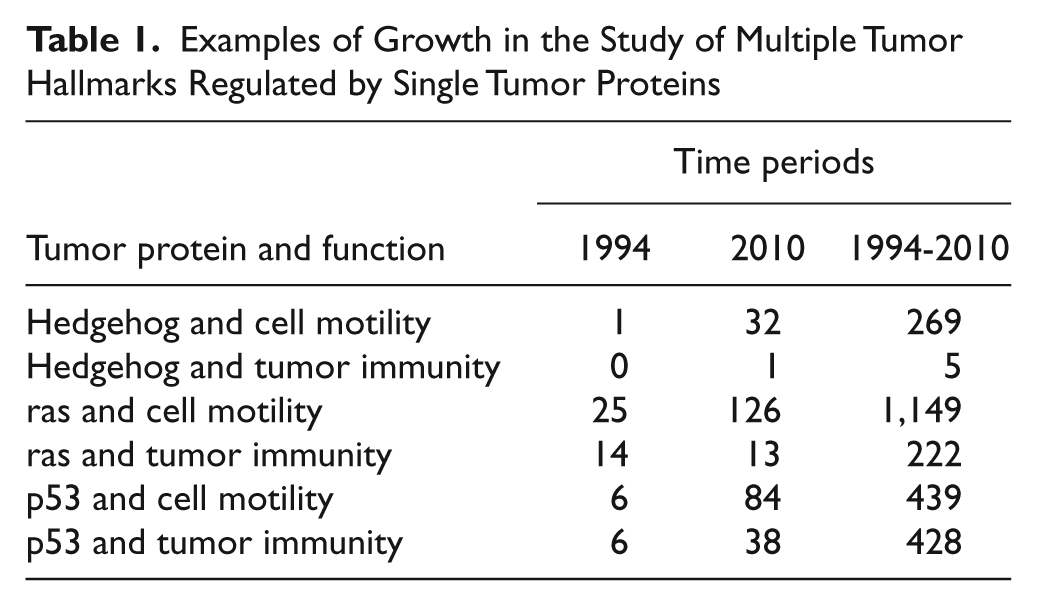
Tumorigenesis is often described as a process whereby cells acquire mutations until the development of a fatal cancer, representing all of the phenotypic hallmarks of the underlying mutations and their myriad, distinct molecular mechanisms.1-3 However, the last 20 years of research has failed to credibly identify tumor mutations that have distinct effector functions. In fact, just the opposite has been the case. In 1993, Taniguchi and colleagues published a linkage between the interferon regulatory factors, first identified as immune system mediators, and the regulation of cell proliferation. 4 In 1994, we published the first link between an established cell cycle regulatory protein and a tumor cell immune function. 5 Since that time, the biomedical research literature has been extensively populated with reports of multifunction oncoproteins and multifunction tumor suppressor proteins. For example, the hedgehog signaling pathway, very credibly linked to cell proliferation, is described in 270 articles as also being relevant to cell motility. Other examples of single proteins linked to multiple hallmarks of tumorigenesis are indicated in Table 1.
Examples of Growth in the Study of Multiple Tumor Hallmarks Regulated by Single Tumor Proteins
Hypothesis
The observation that a single, given oncoprotein or tumor suppressor protein can play a role in multiple, if not all the known, hallmarks of tumorigenesis raises several questions. First, what is the molecular basis for the naturally occurring, sequential patterns of human cancer-inducing mutations,1,6-8 if these mutations do not have distinct functions that facilitate the required, ordered acquisition of hallmarks on the path to a fatal cancer?
We reviewed the inactivated genes in colon cancer, which represents the paradigm for the idea that the sequence of mutations is important for generating a clinically significant cancer. 1 We noticed that the common sequence of inactivation followed the order from larger to smaller coding regions (Table 2). Thus, one possible explanation for the sequence of mutations could be that larger mutagen targets mutate earliest in life and smaller targets, on average, are mutagenized later in life. This process of a temporal ordering of mutations would represent merely the differences in the probabilities of a mutagen altering a larger rather than smaller target.
Classic Paradigm of the Sequence of Genetic Alterations in Colon Cancer
Testing the hypothesis that mutagen target size dictates the order of acquisition of mutations throughout life would require that a large number of tumors be sampled for mutations at very early stages and then repeatedly sampled, as the tumors progress to a fatal course. Eventually, these types of data may be available in sufficient amounts to provide a compelling case that supports or disproves our hypothesis. Currently, only very few cancers, with only relatively few patient samples covering a spectrum of tumor development, have demonstrable sequential orders of mutations.1,6-8 Thus, it is currently impossible to prove or disprove this hypothesis using mutational data representing a sequence of gene inactivations, in many patient samples for many tumors.
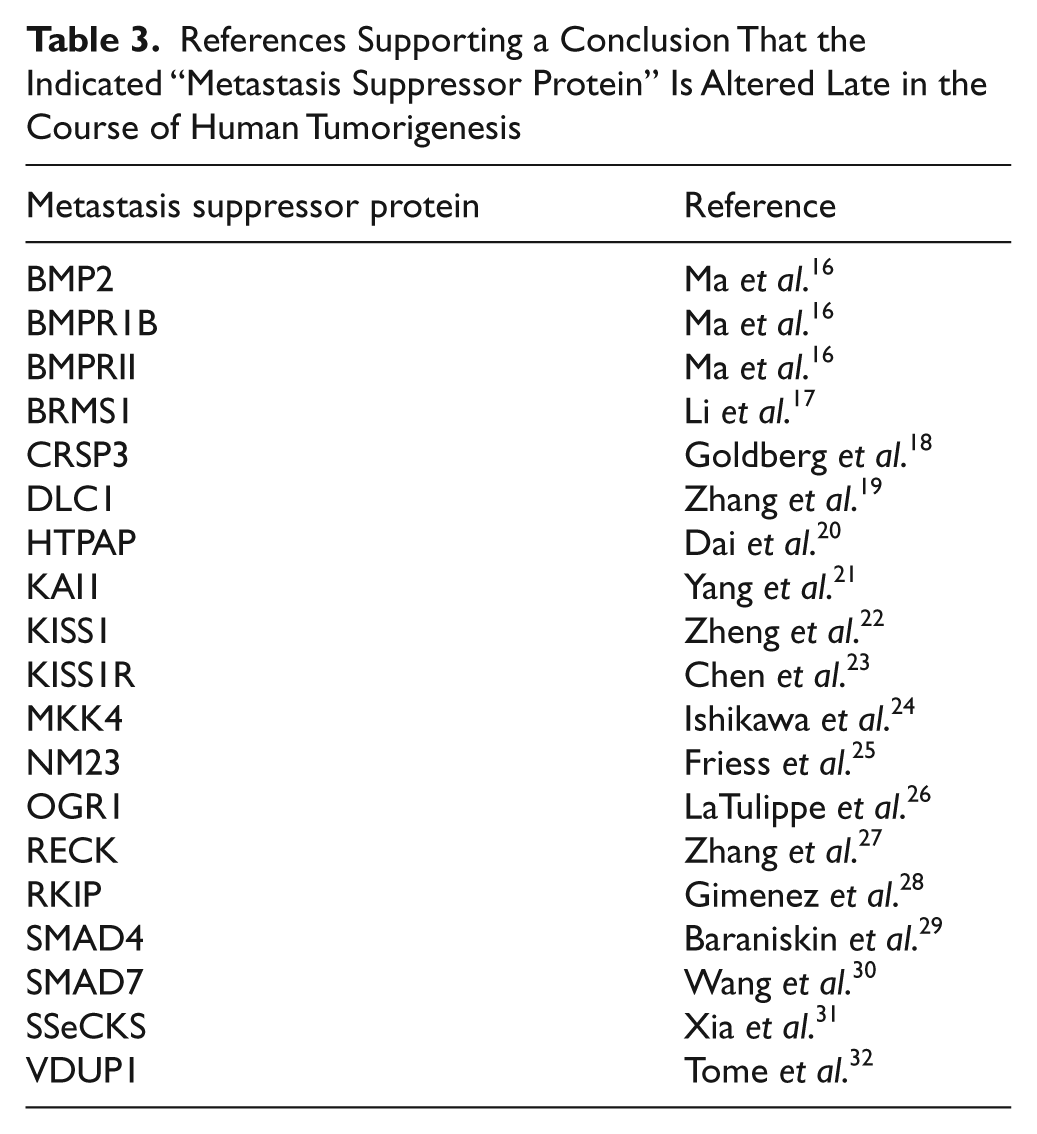
However, by considering proteins that have been accepted as metastasis suppressor proteins, we can address the role of mutagen target size in another manner. Thus, we have assembled 2 sets of proteins inactivated in tumorigenesis: 1) metastasis suppressor proteins (Table 3) and 2) what we will call, for want of a better phrase, “classic tumor suppressor proteins” (Fig. 1 and Suppl. Table S1). We have verified that each member of the set of metastasis suppressor proteins, as first defined by review articles,9-12 is also represented by an original report in the literature as being altered in expression late in tumorigenesis (Table 3), presumably a piece of data important in the initial consideration of each protein as a metastasis suppressor protein.
References Supporting a Conclusion That the Indicated “Metastasis Suppressor Protein” Is Altered Late in the Course of Human Tumorigenesis
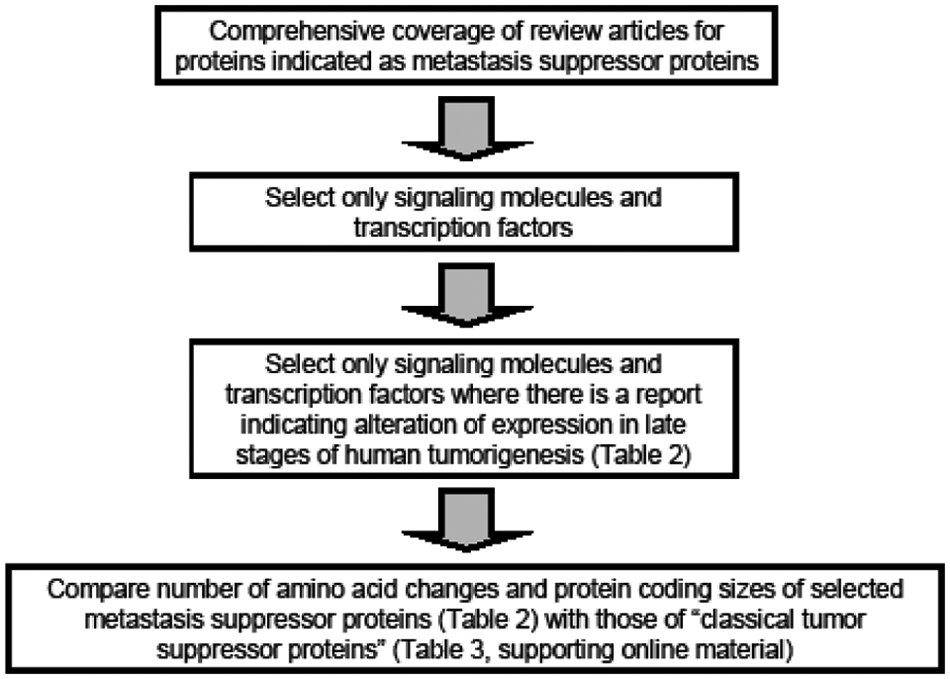
Basis for generating a set of proteins representing metastasis suppressor proteins.
As expected, only several words and phrases, which are closely related to the phrase “metastasis suppressor”, distinguish the 2 sets (Fig. 2A) in the published literature. The 2 sets do not show statistically significant differences in connections to other functions or proteins related to oncogenesis (Fig. 2A), with one exception, the protein, bcl2, linked to the classic tumor suppressor set. This result, or lack of a result, is consistent with the conclusion that metastasis suppressor and tumor suppressor proteins do not differ substantially in their mechanism of function. If we accept the premise that a specific capacity for metastasis would be based on specific intracellular interactions with other proteins involved in oncogenesis, then it is likely that the so-called metastasis suppressor proteins are not specific for metastasis and that the common belief that these proteins specifically prevent metastasis is likely in error.
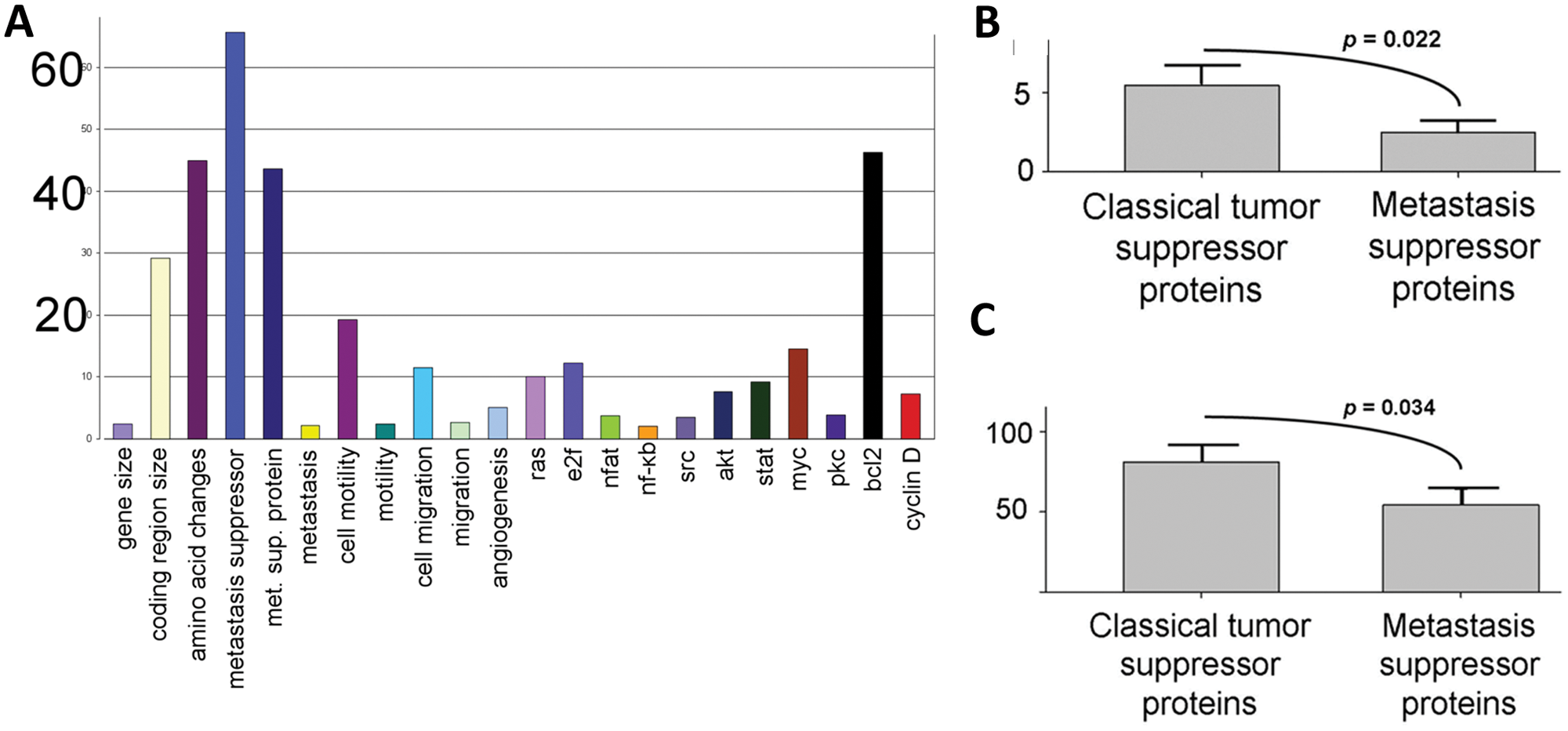
Reciprocal of the P values for the differences in the association of the set of metastasis suppressor proteins with the indicated proteins and functional terms versus the association of the classic tumor suppressor proteins with the indicated proteins and terms. (
Nevertheless, a careful review of the literature verifies that, as a set, the proteins representing the metastasis suppressor set lose their expression in late-stage tumor cells (Table 3). If we accept the imperfect, but undoubtedly approximately correct premise that loss of protein expression is due to mutation of the gene encoding that protein, we can use the 2 indicated sets to ask whether the metastasis suppressor set represents smaller mutagen target sizes, which could explain why it takes a longer time for these genes, on average, to become inactivated. To do this, we tallied the number of amino acids that are known to undergo alterations in the proteins of both sets, which revealed that indeed, the 2 sets have a statistically significant difference in the number of amino acids known to be altered, with the metastasis suppressor set having fewer amino acids that are subject to mutation (Fig. 2A and 2B).
It is possible that the above amino acid alteration results are related to a number of artifacts. Not all of these artifacts can be readily dispensed with, and proof of our hypothesis will inevitably require specific data sets regarding early-stage versus late-stage inactivating mutations, as indicated above. However, the possible role of incomplete research in the number of currently known mutations can be addressed, as follows. We assembled the protein sizes of the 2 groups, accepting the premise that a mutagen target size cannot exceed protein coding region size. Results indicate that the metastasis suppressor genes, on average, have smaller coding sequences than the classic tumor suppressor genes (Fig. 2A and 2C). Thus, there are only 2 parameters that distinguish the metastasis suppressor group from the classic tumor suppressor group, besides the phrase “metastasis suppressor”, and very closely related phrases, and the literature connections to bcl2 (Fig. 2): 1) number of known amino acid changes and 2) protein coding region size, both smaller for the metastasis suppressor group.
Discussion
Despite the advances in genomic approaches to cancer, some time will be needed to obtain enough direct data to convincingly verify the above hypothesis, particularly in view of the fact that a very large amount of data may be needed to observe a favored, temporal order above a background of stochastic variation that involves 4 or 5 mutagen targets, potentially representing small target size variations, and because many early stage tumors are simply not detectable and do not lend themselves to genomic analysis, with a few exceptions, such as colon cancer.
However, at this time, there is no compelling basis for accepting that either the metastasis or classic tumor suppressor group of proteins has an exclusive biochemical function that dictates the appearance of a specific hallmark of cancer, which raises the following question: how do tumor cells acquire different hallmarks during the development of a fatal cancer? The answer in our view is most likely related to quantitative changes in cell signaling levels rather than qualitative changes. For example, inactivation of gene A may lead to rapid cell cycling, whereas inactivation of gene A and gene B would lead to even more rapid cell cycling and to the expression of certain tissue-invading proteases, merely because the genes for the proteases require twice as much effector, signal transduction activity for initiating gene expression, as exemplified by McCawley et al., 13 who report the requirement for sustained MAPK signaling for MMP9 gene expression. The level of signaling activity, as opposed to a signaling pathway being “on” or “off”, effecting a discrete cellular phenotype, has been documented in several cases,13,14 including T-cell function. 15 However, the basic idea of a continuum of increases in signaling leading to the discrete flip of molecular switches that mediate cellular physiology has not been substantially considered by the cancer research community.
Given the above consideration of the level of signaling activity, as opposed to a specific function for different signaling pathways, the order of the inactivation of genes A and B during the course of life would merely reflect the differing number of amino acids, respectively, that could be mutated in a way that leads to inactive tumor suppressor proteins. In an experimental model system, such as a gene-targeted mouse, the order of removing A or B would have no significant effect on the final cell phenotype. Thus, it is possible that metastasis suppressor proteins and classic tumor suppressor proteins have been distinguished by the scientific community first because the metastasis suppressor proteins are more often inactivated late in tumorigenesis. The late inactivation data stimulated hypotheses regarding metastasis specificity. Investigators then effectively tested for only quantitative impacts on the state of cell signaling, which would indeed reveal as impact on metastasis but with no requirement for an exclusive impact on a wholly distinct signaling pathway or set of regulatory proteins.
The hypothesis that ordered mutations of tumor suppressor genes reflect mutagen target sizes leads to several additional expectations. As an order of mutations would be governed by statistical functions, the order of changes would not be invariant, merely common, which was originally reported to be the case for colon cancer. 1 Also, the frequency of a given temporal order would be dependent on the sizes of the genes involved. For example, 2 mutagen targets of a closely related size would frequently vary in temporal order in acquiring mutations, whereas a very large mutagen target would almost always precede a very small mutagen target. Interestingly, almost all, if not all, inherited cancer susceptibility genes are very large tumor suppressor genes, consistent with the idea that these genes represent large mutagen targets, so large that they are mutagenized in embryonic development. Such early mutations would be replicated to constitute tissues that would eventually develop into cells generating gametes.
In many cases, cancer development has included inactivation of genes that provide for many alternatives for inactivation, that is, that represent large target sizes. For example, PTEN can incur a change in one of 20 different amino acids (Suppl. Table S1). If size matters, then genes representing the largest mutagen targets would provide the greatest efficiency in screening for very early changes in tumorigenesis. That is, screening procedures that rely on mutagen targets of very large sizes would have the best chance of revealing the potential for cancer later in life and perhaps the best chance to apply therapies that can be successful only when the tumor burden is low, such as certain immunotherapies. Alternatively, an early occurrence of the low probability hit may make a clinically challenging cancer inevitable, in the sense that the higher probability inactivation of a very large target would be virtually inevitable over the course of life. Thus, early occurrences of low probability hits could justify more diligent anticancer approaches.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the USF Molecular Medicine Faculty Fund.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.