
Abstract
Adenovirus (Ad) early gene 1A 243 residue protein (E1A 243R) possesses a potent transcription-repression function within the N-terminal 80 amino acids (E1A 1-80). We examined the ability of E1A 243R and E1A 1-80 to repress transcription of both an exogenous and the endogenous HER2 promoter in a human breast cancer cell line upregulated for the HER2 proto-oncogene (SK-BR-3). Both moieties repressed HER2 expression by over 90%. When E1A 1-80 was expressed from a nonreplicative Ad vector, levels of expression were lower than anticipated. Addition of nonspecific sequences to the E1A 1-80 C-terminus (E1A 1-80 C+) enhanced its expression 10- to 20-fold. Because “oncogene addiction” suggests that repression of HER2 could kill HER2 upregulated cells, we examined the ability of full-length E1A 243R and E1A 1-80 C+ delivered by an Ad vector to kill HER2 upregulated SK-BR-3 cells. Expression of both E1A 243R and E1A 1-80 C+ killed SK-BR-3 cells but not normal breast cells. E1A 1-80 C+ is a particularly effective killer of SK-BR-3 cells. At 144 h post infection, over 85% of SK-BR-3 cells were killed by a 100 moi of the Ad vector expressing E1A 1-80 C+. As controls, Ad vectors expressing E1A 243R with deletion of all known functional domains or expressing unrelated β-galactosidase had no effect. Three additional human breast cancer cells lines reported to be upregulated for HER2 or another EGF family member (EGFR) were found to be efficiently killed by expression of E1A 1-80 C+, whereas three additional “normal” cell lines (two derived from breast and one from foreskin) were not. The ability of the E1A transcription-repression domain alone to kill HER2 upregulated breast cancer cells has potential for development of therapies for treatment of aggressive human breast cancers and potentially other human cancers that overexpress HER2.
Introduction
Upregulation of the HER2 (ErbB2) proto-oncogene occurs in several types of human cancer, including breast, ovary, and prostate. HER2 upregulation in breast cancer is predictive of aggressive disease with a poor prognosis; 25% to 30% of breast cancers are positive for HER2 upregulation but account for 60% to 80% of breast cancer deaths (reviewed in Moasser 2007 1 ). Of significance, several studies have shown that elevated HER2 levels decrease the susceptibility of cancer cells to chemotherapeutic drugs (reviewed in Moasser 2007 2 ). Further, treatment with siRNAs that inhibit HER2 translation was found to promote growth arrest and apoptosis of breast cancer cell lines. 3 These findings appear to be examples of what has been termed “oncogene addiction.” 4 Several studies with human cancer cell lines have shown that although they may have acquired multiple genetic and epigenetic abnormalities, they can remain highly dependent on the expression of a single oncogene for cell proliferation and survival (reviewed in Weinstein and Joe 2008 5 ). Oncogene addiction has been demonstrated in several mouse model systems including MYC-driven papillomas, lymphomas, and osteosarcomas,6-8 hRAS-driven melanoma, 9 and BCR/ABL-driven leukemia. 10 Interference with the function of the oncogene that drives a specific tumor is a therapeutic approach that has met with some clinical success including targeting BCR-ABL (imatinib), EGRF (gefitinib, erlotinib) and HER2 (Herceptin 11 ) (reviewed in Weinstein and Joe 2008 5 ).
Described here is the use of the adenovirus (Ad) early gene 1A (E1A) repression domain alone to transcriptionally repress the HER2 proto-oncogene in a novel approach to the treatment of HER2 upregulated cancers. The Ad group C (types 2 and 5) E1A oncogene encodes two major proteins of 243 and 289 amino acid residues (243R and 289R) which contain multiple functional domains that interact with key cellular regulatory factors. E1A is involved in diverse functions, including transcriptional activation, induction of cellular DNA synthesis, cell immortalization, cell transformation, and, of particular interest for this study, transcriptional repression. E1A 289R differs from E1A 243R by conserved region 3 (CR3), a 46 amino acid domain unique to 289R that is involved in transcription activation of Ad early genes.12,13 The Ad5 oncogene inhibits the expression of HER2 in rodent and human cell cultures.14-16 However, the full-length Ad E1A oncogene is not a good candidate as a therapy because it possesses, in addition to its transcription repression function, other biological activities which may complicate a medical therapy and could have long-term deleterious effects. Further, other E1A domains interact with several important cellular proteins not associated with its transcriptional repression function, including, for example, Rb, p21, and CtBP, all of which can have profound effects on cell cycle regulation.
Our laboratory first demonstrated that the transcription repression function of the E1A oncogene consists of two critical subdomains that reside solely within the N-terminal 80 amino acids of E1A.17-22 Extensive studies demonstrated that the E1A repression domain (a recombinant protein containing only the N-terminal 80 amino acids) exhibits the same repression function as the entire E1A 243R protein. Single amino acid substitution analysis has demonstrated that there are two E1A N-terminal repression subdomains and has led to a two-step model of E1A repression21,22: first, E1A gains access to repressible promoters by interaction of E1A repression subdomains 1 (amino acids ~1-30) and 2 (amino acids ~48-60) with a promoter-bound cellular partner such as p300; second, the E1A N-terminus (subdomain 1) interacts with TBP (TATA binding protein) and disrupts the TBP/TATA complex thus blocking transcription.23,24 We propose the use of the single functional domain of the multifunctional human Ad early region E1A oncogene to transcriptionally repress HER2 expression and thus negate its function in HER2 mediated breast cancer pathogenesis.
Results
Exogenous and endogenous HER2 promoters are transcriptionally repressed by E1A
MCF-10A cells, which arose by spontaneous immortalization of normal breast epithelial cells, 25 were co-transfected with a luciferase gene (luc) expressed from the HER2 promoter and with varying levels of a plasmid expressing E1A 243R. Fig. 1A demonstrates that expression from an exogenous HER2 promoter is repressed in a dose-dependent manner by E1A 243R.
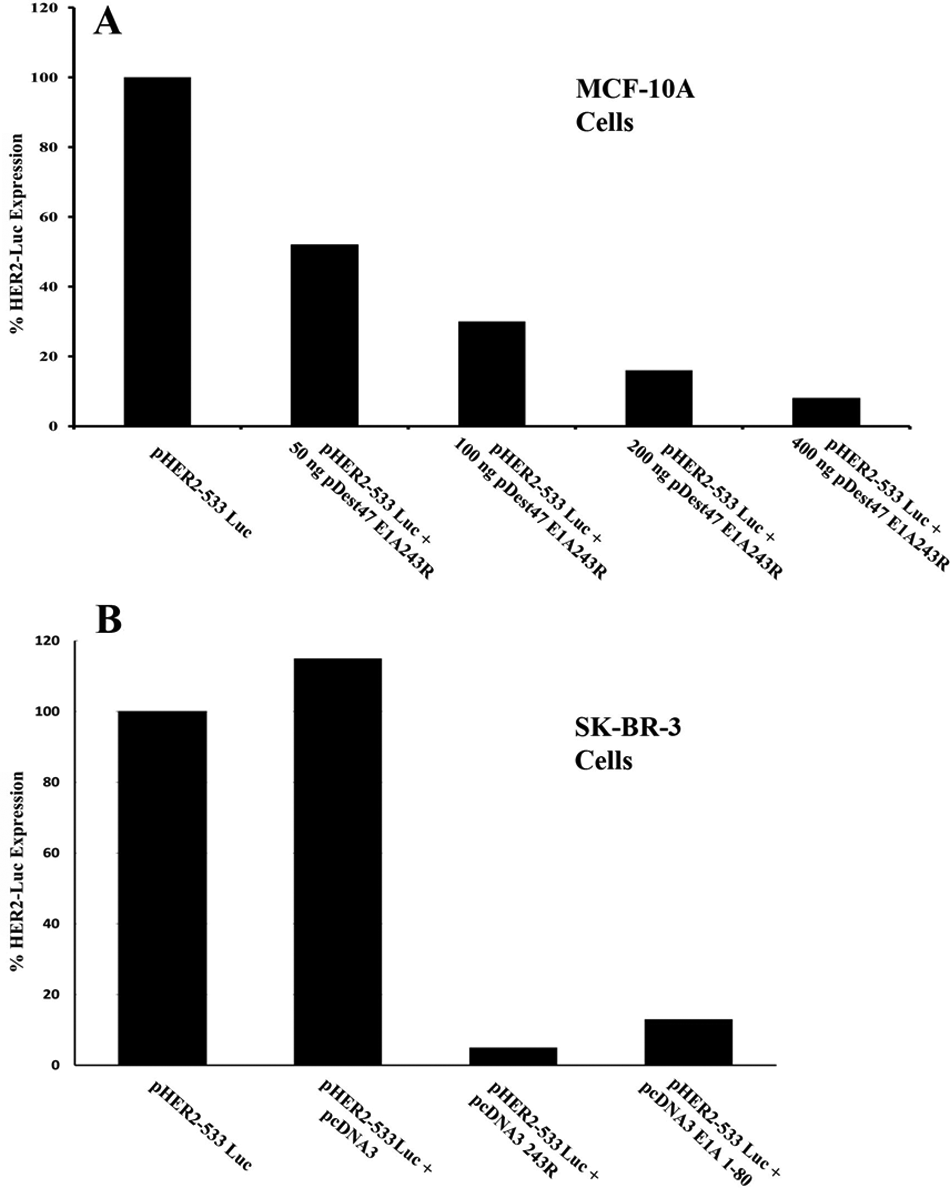
Transcription from an exogenous HER2 promoter is repressed by E1A 243R and E1A 1-80. (
SK-BR-3 cells are frequently used as a model of HER2 upregulation in human breast cancers because SK-BR-3 is amplified 4- to 8-fold for the HER2 gene and SK-BR-3 cells express about 128-fold higher levels of HER2 RNA than normal breast fibroblasts. 26 Therefore we determined whether expression from the transfected exogenous HER2 promoter could be repressed by E1A 243R in an environment of very high exogenous and endogenous HER2 expression. As shown in Figure 1B, when E1A 243R is expressed in human SK-BR-3 cells, it is able to repress by over 15-fold, a co-transfected “exogenous” HER2 promoter driving a luciferase gene. As anticipated from our earlier studies, the E1A repression domain alone (E1A 1-80) efficiently represses transcription from the HER2 promoter construct. This confirms that the E1A repression domain alone is capable of repressing the HER2 promoter in vivo and sets the stage for testing the ability of the repression domain to repress endogenous HER2 expression in upregulated human cancer cells.
To determine whether the endogenous HER2 promoter can be efficiently repressed in SK-BR-3 cells, E1A 243R and E1A 1-80 were cloned into a replication-deficient Ad vector lacking the E1A, E1B and E3 genes (AdCMV/V5; Invitrogen, Carlsbad, CA). In this vector, the cloned E1A 243R and the E1A 1-80 repression domain are expressed from the strong CMV promoter. As shown in Figure 2, when E1A 243R is expressed from AdCMV in SK-BR-3 cells at either 30 or 300 moi, HER2 expression is reduced over 80% by 36 h post infection (PI) as quantitated by real-time RT-PCR. Expression of E1A 1-80 from this Ad vector also repressed expression of HER2 but at a level that was less than anticipated (data not shown).
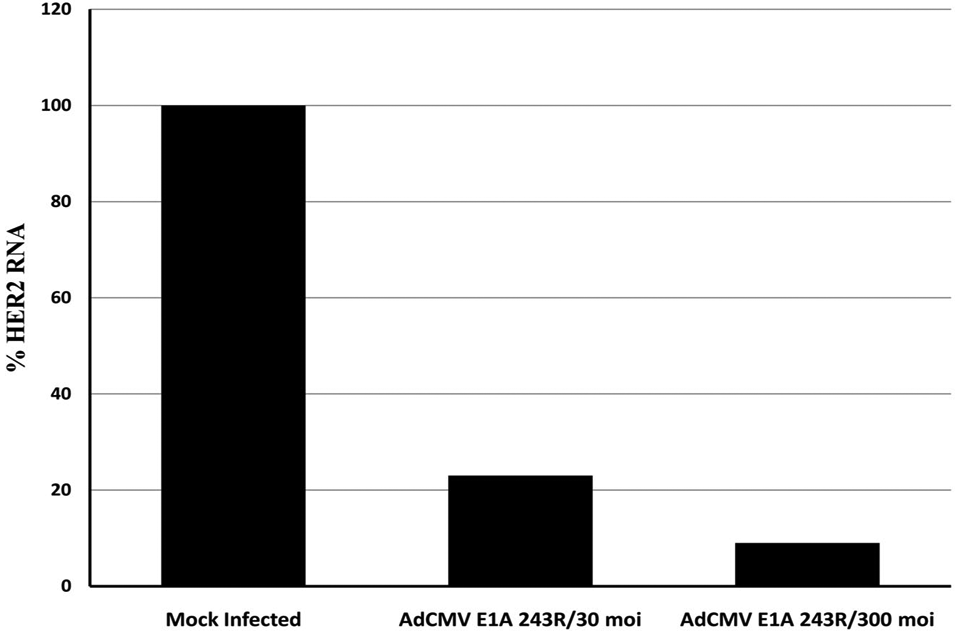
E1A 243R, when expressed from an Ad vector, is able to repress the transcription of the endogenous HER2 promoter in SK-BR-3 human breast cancer cells. Cells were infected with 30 or 300 moi of AdCMV E1A 243R and harvested at 36 h PI. Expression of HER2 was measured by quantitative RT-PCR. Data are from a representative experiment.
Modification of the C-terminus of E1A 1-80 dramatically increases its expression
The difference between AdCMV E1A 243R and AdCMV E1A 1-80 is the 163 amino acids removed from the C-terminus of E1A 243R. In an attempt to either stabilize the E1A repression domain or increase its transcription, we elected to add additional sequences to the E1A 1-80 N-terminus. Re-cloning E1A 1-80 without a stop codon into the AdCMV/V5 vector provided a facile way to accomplish this task. This adds 39 additional amino acids to the E1A N-terminal repression domain (referred to as Ad E1A 1-80 C+). These sequences although containing a V5 epitope were not anticipated to provide any specific structure; 23 nonpolar, 5 acidic, 5 basic, 3 aromatic, and 3 polar residues are included in the additional sequences.
Much more (~10- to 20-fold) E1A repression domain protein, as detected by polyclonal antibody directed against E1A CR1, is produced when A549 cells are infected with either 30 or 300 moi (multiplicity of infection) of Ad E1A 1-80 C+ than when infected with Ad E1A 1-80 (see Fig. 3A). In Figure 3A, the apparent size of the products does not reflect the 39 additional amino acids present in Ad E1A 1-80 C+, but sequence analysis shows them to be present. Anomalous apparent size by SDS PAGE is commonly observed with E1A proteins.
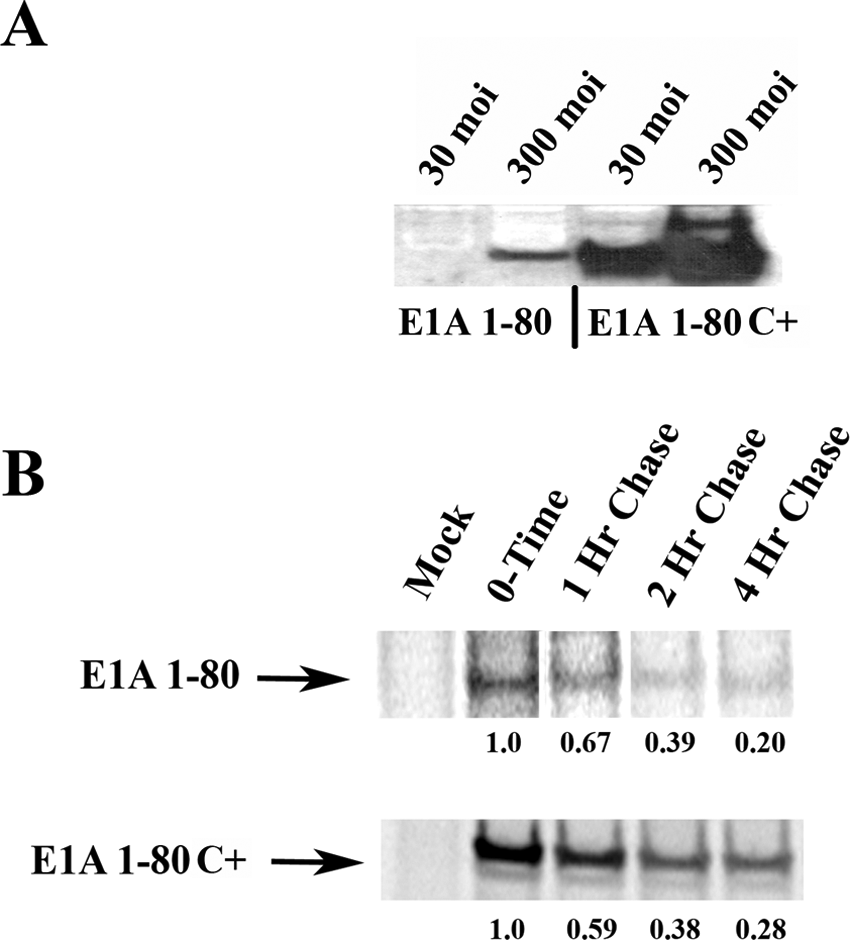
E1A 1-80 modified at its C-terminus (E1A 1-80C+) is expressed from an Ad vector at high levels. (
Although there is clearly much more protein produced from Ad E1A 1-80 C+ as compared to Ad E1A 1-80, the reason for this is not apparent. An obvious question is whether the transcript is stabilized or whether transcription is enhanced. Figure 3B shows the results of parallel pulse-chase experiments using E1A 1-80 or E1A 1-80 C+. As can be seen, the total amount of protein produced by infection with AdCMV E1A 1-80 C+ is more than that produced by infection with AdCMV E1A 1-80, but the rate of protein turnover, which reflects mRNA half-life, is about the same. These findings suggest that the rate of transcription and not product stability accounts for the increased levels of E1A protein as seen in Figure 3A. However, we have not excluded the possibility that translational control could play a role.
Expression of the Ad E1A repression domain kills SK-BR-3 cells
To determine whether transcription-repression of the endogenous HER2 promoter would interfere with the growth of HER2 upregulated human breast cancer cells, SK-BR-3 cells were infected with varying levels of Ad vector expressing either E1A 243R or E1A 1-80 C+. As controls for potential toxicity from Ad vector infection, two additional Ad vectors were used: E1A 243R dl1101/dl1108/dl135 23 which is deficient in known E1A 243R functional domains, and LacZ, an unrelated gene, were expressed from the parental vector AdCMV/V5 in parallel wells. Cell killing was measured in replicative plates every 48 h by a cell viability colorimetric assay, as described in the Materials and Methods section.
When SK-BR-3 cells were infected at 30 moi (Fig. 4A, first panel), both AdCMV E1A 243R and AdCMV E1A 1-80 C+ clearly exhibited cell killing by 96 h PI. By 192 h PI, AdCMV E1A 1-80 C+ showed pronounced cell killing (~75%) compared to AdCMV E1A 243R (~50%) relative to mock infected or cells infected with the control Ad vectors. When the normal human breast cell line HS 579.Mg was infected at 30 moi with the same panel of Ad vectors (Fig. 4B, first panel), no difference between AdCMV 243R or AdCMV E1A 1-80 C+ could be detected relative to mock infected or the two Ad vector controls.
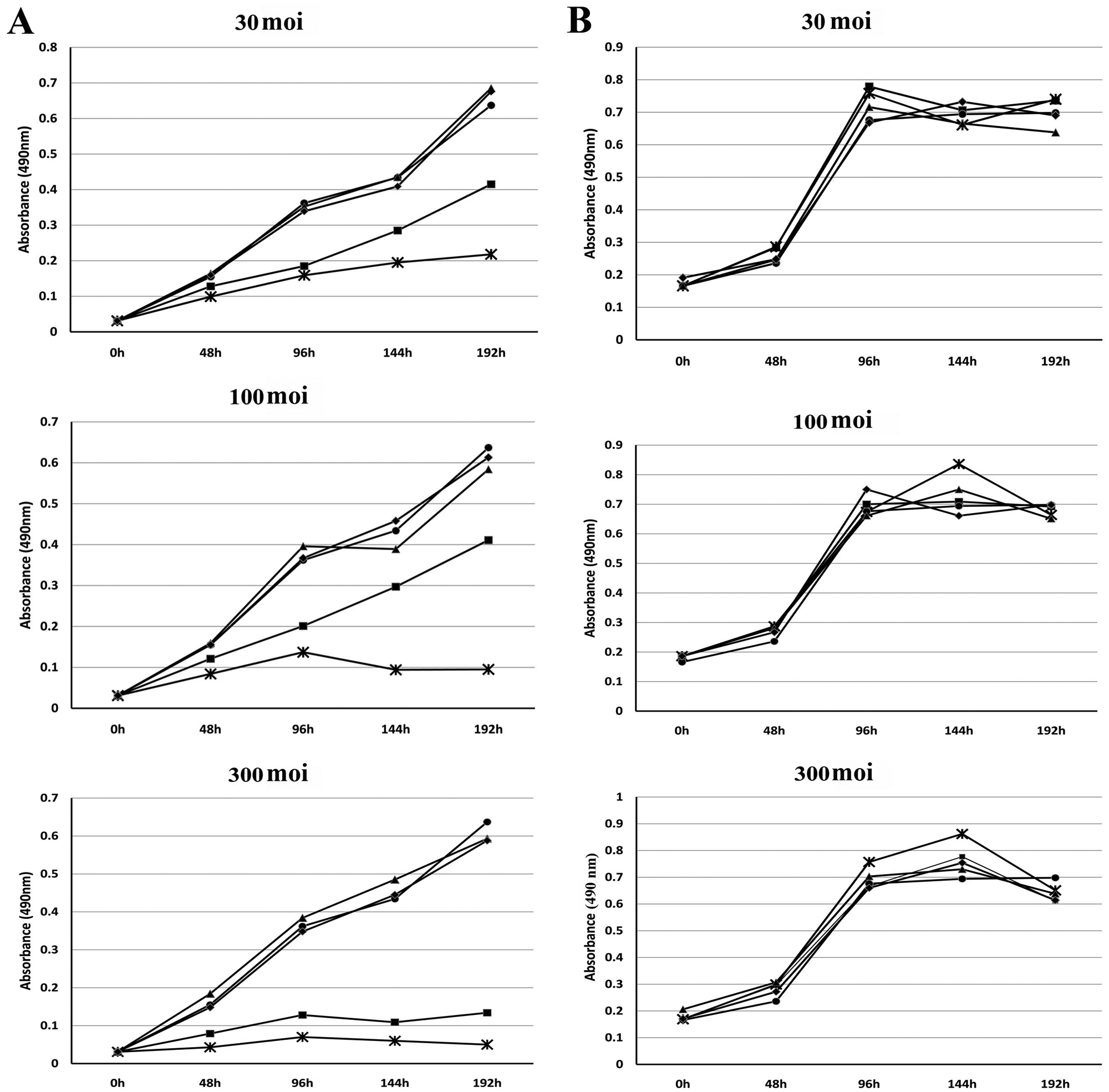
(
At 100 moi (Fig. 4A, second panel), there was a greater difference between the cell-killing efficiencies of AdCMV E1A 243R and AdCMV E1A 1-80 C+. Again, SK-BR-3 cell killing by AdCMV E1A 243R and AdCMV E1A 1-80 C+ is clearly apparent by 96 h PI. However by 144 or 192 h PI, cell killing by AdCMV E1A 1-80 C+ was over 85% whereas cell killing by AdCMV E1A 243R was not substantially different from that observed with infections at 30 moi. Control Ad vectors did not exhibit substantial differences from mock infected. Again, when HS 579.Mg cells were infected with the Ad vectors (Fig. 4B, second panel), no significant cell killing by AdCMV E1A 243R or AdCMV E1A 1-80 C+ could be detected.
At 300 moi (Fig. 4A, third panel), SK-BR-3 cells infected with both AdCMV E1A 243R and AdCMV E1A 1-80 C+ showed very substantial cell killing. Significantly, even at this high moi, the controls exhibited no cell death when compared with mock infected cells. HS 579.Mg cells (Fig. 4B, third panel) infected with AdCMV 243R or AdCMV 1-80 C+ did not exhibit significant cell killing.
Expression of the Ad E1A repression domain kills other breast cancer cells but not normal cells
To evaluate the generality of these observations, we tested the ability of E1A 1-80C+ to kill three additional human breast cancer cell lines (MB-453, MB-468, and MB-231) and three additional “normal” cell lines; two human breast cell lines (MCF 10A and MCF 12F) and a human foreskin fibroblast cell line (HS68). The human breast cancer cell line MB-453 has been reported to over-express HER2 27 ; MB-468 expresses the EGF receptor as does MB-231, which has been used as a model for EGF expression in cancer cells (for example, see Fan et al. 1998 28 ). All three of the human breast cancer cells were efficiently killed when E1A 1-80C+ was expressed at a moi of 300; the three normal cells lines were unaffected by expression of E1A 1-80C+ (see Fig. 5).
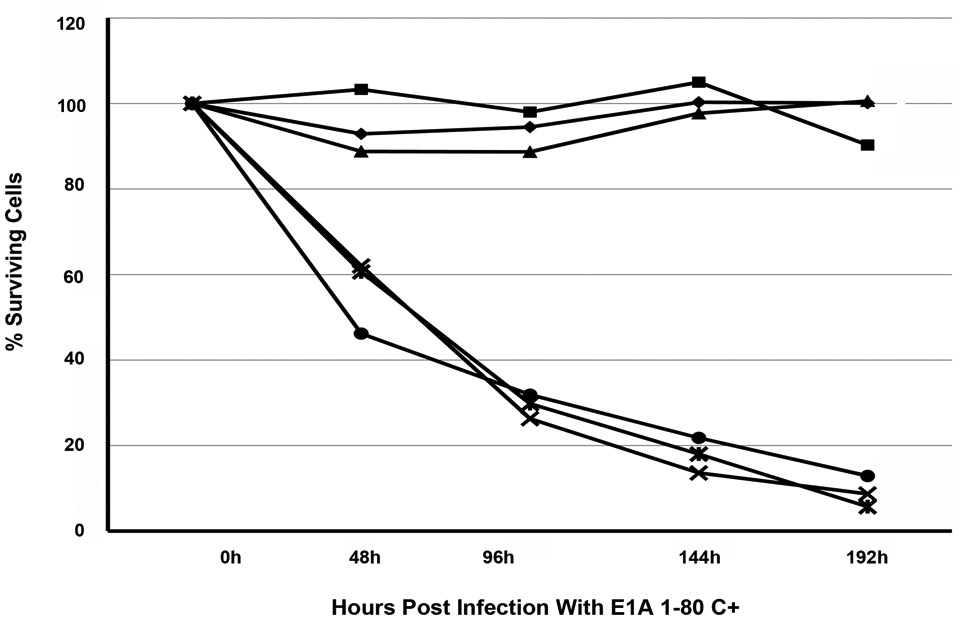
Human breast cancer cells, MB-468 (■–■), MB-453 (◆–◆) and MB-231 (▲–▲) or normal human cells, MCF 10A (•–•), MCF 12F (
To summarize, expression of the E1A repression-domain leads to HER2 downregulation in SK-BR-3 cells and results in cell death. E1A 1-80 C+ proved to be a significantly more efficient killer of SK-BR-3 cells at low or moderate moi than E1A 243R. At all moi’s tested, neither E1A 243R or E1A 1-80 C+ had a significant effect on the normal HS 579.Mg breast cell line, and no cell death could be attributed to infection by Ad vectors alone. Expression of E1A 1-80 C+ also resulted in efficient cell killing of three additional breast cancer cells lines expressing HER2 or another EGF receptor family member. Two other “normal” breast cell lines and a normal human foreskin fibroblast were not killed by expression of E1A 1-80 C+.
Discussion
It is clear from the data presented here that E1A 243R, and more importantly the E1A 1-80 repression domain alone, can efficiently repress the expression of HER2 and when expressed in the HER2 upregulated SK-BR-3 breast cancer cell line leads to cell death. SK-BR-3 cells were used as a prototype for human breast cancers dependent upon upregulated HER2 oncogene for growth and survival. The fact that expression of the E1A transcription-repression domain efficiently kills these cells is consistent with the “oncogene addiction” hypothesis. 4 This view holds that, at least in some cancers, once a tumor cell becomes dependent on the continued expression of an oncogene, interference with the expression or function of that oncogene can lead to a return to a normal phenotype or to cell death. Oncogenesis, however, is a continuing process that accumulates genetic and epigenetic abnormalities and it is entirely possible that a cancer cell may “evolve” beyond dependence on the original oncogene (reviewed in Luo et al. 29 ). Therefore, if therapy targeting a tumor exhibiting an oncogene addiction is to succeed without the development of resistant tumors, it must be very effective or target more than one aspect of oncogene function. For example, as many as one-third of patients having advanced disease treated with herceptin fail to respond and further many of those who initially respond show progression of their disease within one year of treatment.30,31 More recently, there is evidence that HER2 upregulation results in an increase in the number of malignant stem cells which are “tumor initiating cells” and may contribute to the failure of treatment. 32 This underscores the potential value of E1A 1-80 to transcriptionally repress HER2 for the treatment of breast cancer.
Transcription repression of the biosynthesis of a specific oncoprotein, e.g., HER2 protein, is a strategy that has not been applied as an antioncogene therapy, but the SK-BR-3 cell killing shown here demonstrates its potential. Herceptin, a humanized monoclonal antibody directed against the HER2 receptor, has proven its utility, but even a small percentage of unblocked receptors can allow the HER2 signal cascade to stimulate cancer cell growth, thus allowing the continued risk of the development of herceptin-resistant cells. A multidrug approach targeting different aspects of the same cancer promoting mechanism is more likely to succeed, as the failure of one approach is not likely to affect the other. For example, herceptin used in concert with an E1A repressor of HER2 transcription may produce a superior inhibition of HER2 function overall, with less likelihood for the development of resistant tumors.
The relatively small E1A 1-80 repression domain has potential for development into a successful therapy, as shown by its ability to kill at least four human breast cancer cells lines while not killing four cell lines derived from “normal” cells. Other than transcriptional repression, E1A 1-80 does not possess the complicating functions of the multifunctional E1A 243R oncoprotein. Its size does not preclude its modification by peptide mimetics nor delivery by methods other than Ad vectors. It appears from the studies presented here to be a more potent killer of SK-BR-3 cells than E1A 243R, probably because it is more efficiently transcribed in its modified E1A 1-80 C+ form. Further examination is needed to access the practicality of using of the E1A transcription-repression domain as a therapy for the treatment of aggressive HER2 upregulated human breast cancers.
Materials and Methods
Plasmids and transfections
pHER2-533 expressing the luciferase gene driven by the HER2 promoter 33 was used as reporter. E1A 243R was PCR cloned into the expression vector pDest47 (Invitrogen), or E1A 243R and E1A 1-80 were PCR cloned into pcDNA3 (Invitrogen) using appropriate primers; pLE2 dl320, a plasmid containing genomic E1A with a splice-point mutation causing only 243R to be transcribed was used as template.23,34 MCF-10A (ATCC) cells were transfected in 24 well plates with a total of 500 ng of DNA using lipofectamine (Invitrogen) and the manufacturer’s guidelines. SK-BR-3 (ATCC) cells in 60 mm2 cell culture dishes were transfected with a total of 1 µg of DNA using Fugene (Roche, Indianapolis, IN) and the manufacturer’s guidelines. Cells were harvested 48 h post transfection and luciferase gene expression quantified after normalization against expression of a co-transfected non-E1A repressible RTL-luc using a Dual-Luciferase assay kit (Promega, Madison, WI) and a Turner Design luminometer.
Adenovirus vectors
E1A 243R, E1A 243R mutants with stop codons, and E1A 1-80 with or without a stop codon were cloned into the Gateway (Invitrogen) entry vector pENTR/SD/D-TOPO following the manufacturer’s instructions. After sequence confirmation, E1A inserts were transferred into pAd/CMV/V5 DEST (Invitrogen). E1A 243R, the triple mutant E1A 243R dl1101/dl1108/dl1135 or E1A 1-80 cloned into the entry vector with a stop codon transcribes their respective E1A moiety under the control of the CMV promoter. E1A 1-80 cloned without a stop codon transcribes E1A 1-80 with an additional 39 amino acids at its C-terminus (AdCMV E1A 1-80 C+). The pAd CMV plasmids were digested with PacI and transfected into 293A cells (Invitrogen). Resultant Ad vectors were amplified, purified on CsCl density gradients and titrated by plaque assay as described. 35
HER2 RNA transcription-repression assays
SK-BR-3 cells, about 60% confluent, in 6 well cell culture plates were infected in 2 mL of DME/10% FBS with AdCMV E1A 243R or AdCMV E1A 1-80 at 37°C in a humidified incubator. After 1 h, 2 mL of complete medium was added and incubation continued for an additional 36 to 48 h and cells harvested by scraping into 1 mL of cold PBS. Cells were washed once in PBS and RNA isolated using an RNA Easy kit (QIAGEN, Santa Clarita, CA). cDNA was prepared using a High Capacity cDNA Archive kit (Applied Biosystems, Carlsbad, CA). Levels of HER2 cDNA were measured by quantitative RT-PCR with a HER2 specific TaqMan probe-set (Applied Biosystems) using an Opticon 2 real-time PCR instrument (Bio-Rad, Hercules, CA).
Western blots and pulse-chase analysis
For Western blots, A549 cells in 60 mm2 cell culture dishes were infected with the indicated amounts of AdCMV E1A 1-80 or AdCMV E1A 1-80C+. Cells were harvested 48 h post infection and subjected to SDS PAGE. Electroblots were probed by antibody specific for the E1A CR1 domain (PD1). 21 Immunoblots were developed using a Super Signal Western Blot Chemo-luminescence Kit (Pierce, Rockford, IL).
For pulse-chase experiments, A549 cells in replicate 10 cm2 cell culture plates were mock infected or infected with 100 moi of AdCMV vector expressing E1A 243R, E1A 1-80, or E1A 1-80C+. At 20 h after infection, cells were washed in PBS and starved for 1 h in DME lacking cysteine/methionine and containing 10% dialyzed FBS. Cells were then labeled for 90 min with 100 µCi/plate of 35 S-labeled methionine/cysteine (Tran-S label; MP Biochemicals, Costa Mesa, CA) and then washed and chased for various times with complete DME/10% FBS.
Cell proliferation assays
Human breast cancer and normal human cells (2000/well) were plated in 100 µL of DME/ 10% FBS into replicate 96 well cell culture plates. Medium was removed 2 h after plating and cells infected with 100 µL of DME/10% FBS containing the indicated amount of Ad vector. Cell culture medium was exchanged for fresh medium (without Ad vector) at 24 h post infection. Cells were fed at 48, 96, 144, and 192 h post infection by replacing the cell culture medium with 100 µL of fresh media. Replicate plates were assayed at 48, 96, 144, and 192 h post infection for cell viability using a Cell Titer 96 cell proliferation assay kit (Promega). In this assay, viable cells bioreduce a MTS tetrazolium compound to a colored formazan product that is soluble in cell culture medium. Cell viability was quantitated using a Thermo Max microplate reader (Molecular Devices, Sunnyvale, CA) at 490 nm.
Footnotes
Acknowledgements
We thank C.E. Mulhall for valuable editorial assistance.
The authors declared no conflicts of interest with respect to the authorship and/or publication of the article.
This work was supported by Research Career AI-04739 and Public Health Service Grant 5R01CA29561 to MG and by a generous gift from the Saint Louis University Cancer Center and with support from Frank and Ruth Stroble.