
Abstract
The vascular network delivers oxygen (O2) and nutrients to all cells within the body. It is therefore not surprising that O2 availability serves as a primary regulator of this complex organ. Most transcriptional responses to low O2 are mediated by hypoxia-inducible factors (HIFs), highly conserved transcription factors that control the expression of numerous angiogenic, metabolic, and cell cycle genes. Accordingly, the HIF pathway is currently viewed as a master regulator of angiogenesis. HIF modulation could provide therapeutic benefit for a wide array of pathologies, including cancer, ischemic heart disease, peripheral artery disease, wound healing, and neovascular eye diseases. Hypoxia promotes vessel growth by upregulating multiple pro-angiogenic pathways that mediate key aspects of endothelial, stromal, and vascular support cell biology. Interestingly, recent studies show that hypoxia influences additional aspects of angiogenesis, including vessel patterning, maturation, and function. Through extensive research, the integral role of hypoxia and HIF signaling in human disease is becoming increasingly clear. Consequently, a thorough understanding of how hypoxia regulates angiogenesis through an ever-expanding number of pathways in multiple cell types will be essential for the identification of new therapeutic targets and modalities.
Hypoxia
All eukaryotic organisms rely on oxygen (O2) to support oxidative phosphorylation for efficient adenosine triphosphate (ATP) production. Therefore, a constant O2 supply, maintained by the vascular system in mammals, is critical for proper tissue development, homeostasis, and function. 1 Tissue oxygenation is governed by a balance between O2 supply, delivered by the vasculature, and demand generated by metabolic outputs of tissues. 2 Hypoxia emerges when changes in the supply or demand occur and is common in both normal mammalian development and human disease. Vascular dysfunction due to vessel occlusion or rupture can cause decreased O2 delivery and is a pathogenic driver in diabetic retinopathy, peripheral artery disease, and ischemic heart disease. 2 In contrast, rapid cellular division during embryonic development or tumor growth can enhance O2 demand due to increased metabolism, causing localized hypoxia. 3 In biological systems, it is important to consider that hypoxia is relative, as the normal physiological O2 concentration varies greatly from tissue to tissue. For example, arterial blood has a normal pO2 of 14%, the myocardium has a pO2 of 10%, and most tissues have a pO2 of ~5%. 4 However, cartilage, bone marrow, and thymus have domains that are naturally at or below 1% O2.5-7 These observations demonstrate that hypoxia is not always a pathological entity and there is increasing evidence that it is an important component of some cellular niches, particularly those of stem and progenitor cells. 8 Nonetheless, low O2 and its molecular sequelae play a critical role in the pathogenesis of a broad array of human diseases, especially those in which the vasculature is a component.
The Major Oxygen Homeostasis Regulators: Hypoxia-Inducible Factors (HIFs)
First identified based on its ability to regulate erythropoietin (EPO) expression, the founding member of the hypoxia-inducible factor (HIF) family, HIF-1α, regulates a broad array of genes in response to O2 deprivation.9-11 The HIF proteins belong to the bHLH-PAS (basic helix loop helix-Per/ARNT/Sim) family of proteins 12 and consist of 3 O2-regulated α-subunits, HIF-1α, HIF-2α, and HIF-3α, and a constitutively expressed β-subunit of the Aryl hydrocarbon nuclear translocator family, including Arnt, Arnt2, and Arnt3 (Figure 1). The HIF-α subunits are structurally homologous, containing an N-terminal bHLH domain that mediates DNA binding and specificity. The HLH and PAS domains mediate heterodimerization with the transcriptional partner Arnt. Although the Arnt family members are all structurally homologous, not all Arnts promote adaptive responses to hypoxia. 13 Arnt3, known as BMAL, is an essential regulator of circadian rhythms, whereas Arnt2 is primarily involved in neural development, where it complexes with the single-minded homologue1 (SIM1). Arnt2 mediates some transcriptional responses to hypoxia in the CNS, although Arnt is the primary HIF-β involved in the hypoxic response. 14
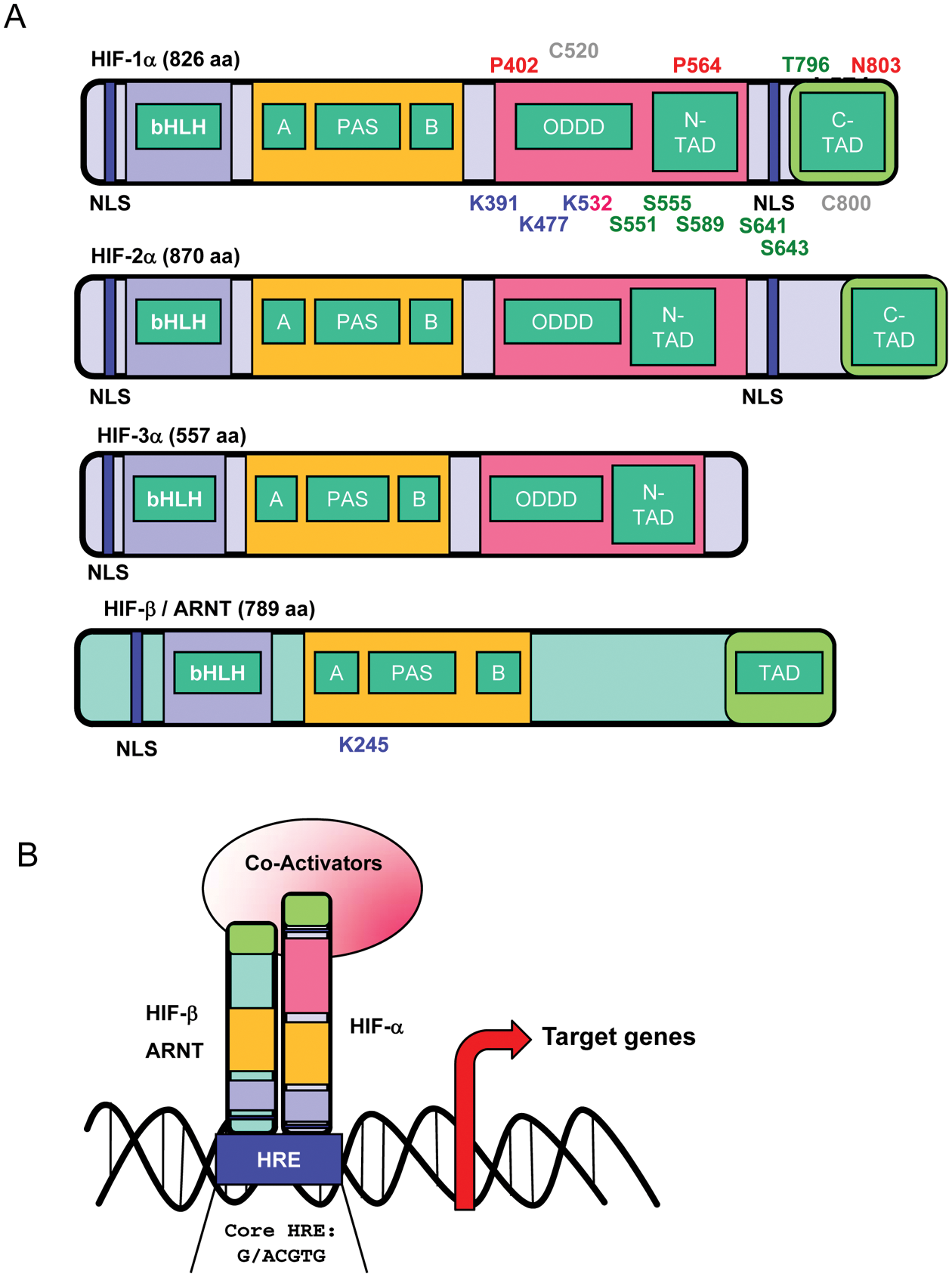
Schematic representation of HIF-α and HIF-β structures and DNA binding. (A) Hypoxia-inducible factors (HIFs) are heterodimers composed of 2 subunits: one α (HIF-1α, HIF-2α, HIF-3α) and one β (HIF-β/ARNT). Each subunit contains different functional domains implicated in nuclear localization (NLS), DNA binding (bHLH/PAS), dimerization (bHLH/PAS), protein stability (ODDD/N-TAD), cofactor interactions (N-TAD/C-TAD), and transcriptional activity (N-TAD/C-TAD). NLS = nuclear localization signal; bHLH = basic helix loop helix domain; PAS = Per-ARNT-Sim motif; ODDD = oxygen-dependent degradation domain; N-TAD = N-terminal transactivation domain; C-TAD = C-terminal transactivation domain. For simplicity, only relevant amino acid residues for HIF-1α and HIF-β are represented. However, distinct residues have been identified for HIF-2α. Hydroxylated residues (red), phosphorylated residues (green), sumoylated residues (blue), nitrosylated residues (gray), and acetylated residues (pink). (B) Active HIF complex binds specific hypoxia-responsive elements (HRE, consensus sequence: G/ACGTG) in the promoters of target genes with the basic residues near the N-terminal terminus of each subunit and induces transcription with co-activators bound through the transactivation domains (TAD) of each protein.
HIF pathway activity is regulated by the prolyl hydroxylase enzymes (PHD1-3), members of the Fe(II) and 2-oxoglutarate-dependent dioxygenase family. 15 In O2-replete conditions, PHD enzymes hydroxylate HIF-α on 2 conserved proline residues located within the HIF-α O2-dependent degradation domain (ODDD) in an oxygen and 2-oxoglutarate-dependent manner. Proline hydroxylation initiates binding to the von Hippel–Lindau E3 ubiquitin ligase complex that ubiquitinates HIF-α, targeting it for proteasomal degradation (Figure 2). Under hypoxia, the PHD substrate O2 and cofactor 2-oxoglutarate become limiting, attenuating HIF-α hydroxylation and resulting in HIF-α accumulation. 16 In this manner, the PHD enzymes may serve as direct cellular O2 sensors. 16 Once HIF-α accumulates in the cell, it subsequently translocates into the nucleus, where it binds to Arnt and forms a transcriptional complex with p300 and CBP through interactions with the HIF-α N- and C-terminal transactivation domain (N-, C-TAD).17,18 HIF-α/Arnt binds to hypoxia response elements (HREs) with the consensus sequence (G/ACGTG) in the promoters and enhancers of target genes 19 (Figures 1-2). HIF-α activity and stability are regulated via additional posttranslational modifications. For instance, factor-inhibiting HIF-1α (FIH-1), another Fe(II) and 2-oxoglutarate-dependent dioxygenase, hydroxylates a conserved asparaginyl residue in the C-TAD of HIF-α, which inhibits binding to p300 and CBP. 20 HIF-α is also modified by acetylation, 21 phosphorylation,22,23 sumoylation,24-26 and s-nitrosylation.27,28 For instance, the p300/CBP-associated factor (PCAF) acetylates HIF-1α under hypoxia, which promotes HIF-1α accumulation. 29 This modification is balanced by Sirtuin1 (Sirt1), which deacetylates both HIF-1α and HIF-2α. Interestingly, Sirt1-mediated deacetylation of HIF-1α inhibits its transcriptional activity, whereas Sirt1-mediated deacetylation of HIF-2α potentiates its transcriptional activity.29,30 Phosphorylation can also positively and negatively regulate HIF activity and stability. 31 SUMO-1 sumoylates Arnt and HIF-1α on conserved lysine residues, which may influence the transcriptional activity of HIF-1α. 24 S-nitrosylation (on cysteine 522 and 800) increases the binding of HIF-1α with co-activators CBP and p300, increasing HIF-1α transcriptional activity 27 (Figure 1). The functional consequences of these modifications are not yet fully understood and are still under debate, although it is clear that the HIFs are extensively posttranslationally modified.
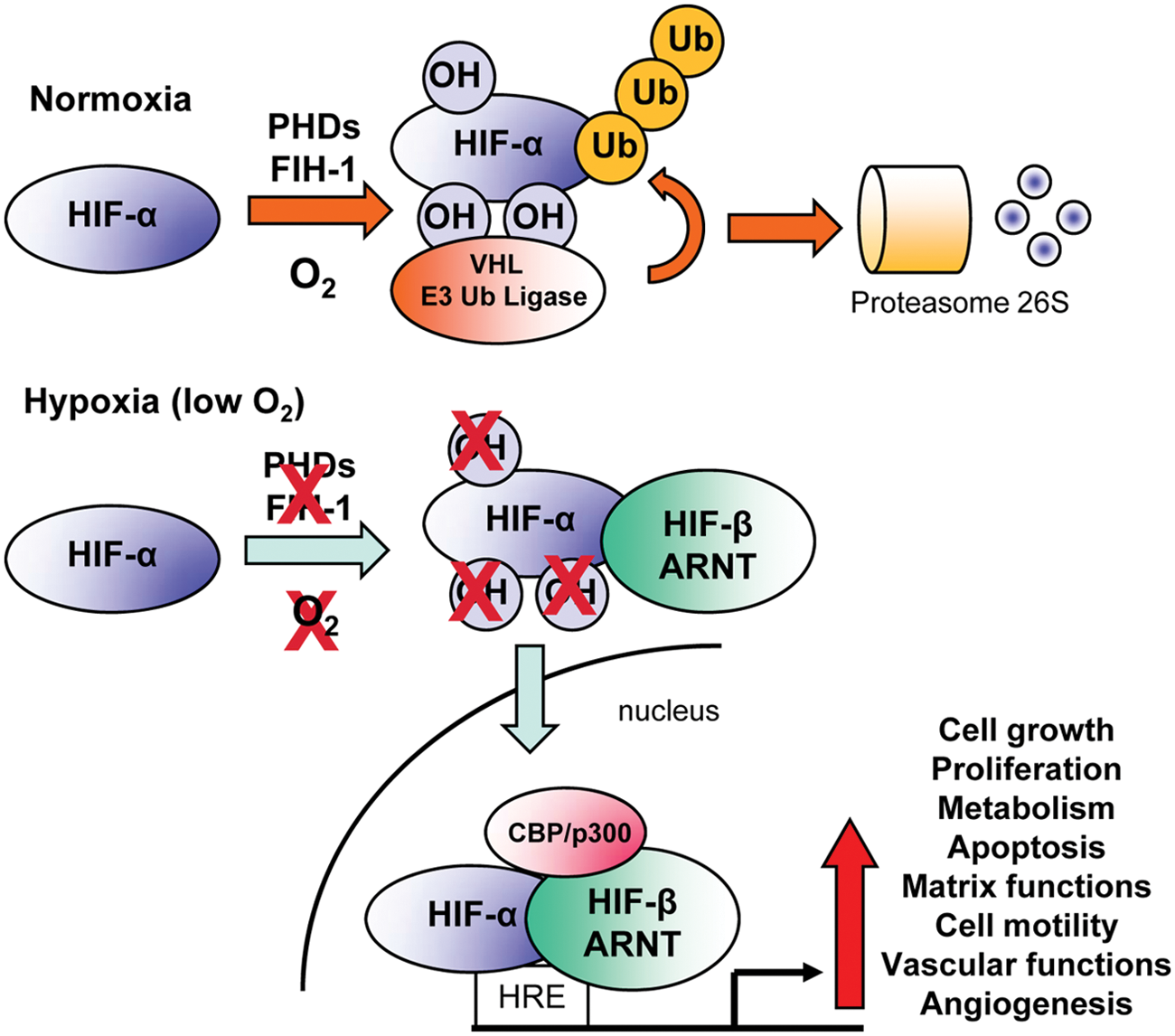
Regulation of hypoxia-inducible factor (HIF) activity. Under normoxic conditions, HIF-α subunits are polyubiquitinated at 2 proline residues within the oxygen-dependent degradation domain (ODDD) by a family of enzymes known as prolyl hydroxylases (PHDs). This promotes recognition by the VHL E3 ubiquitin ligase complex and subsequent degradation of HIF-α via the 26S proteasome. In addition, hydroxylation of a C-terminal asparagine residue of HIF-α by factor-inhibiting HIF-1 (FIH-1) prevents binding of cofactors required for HIF activity. Hypoxia inhibits the activity of the PHD and FIH-1 enzymes, allowing HIF-α proteins to escape recognition by VHL, be stabilized, and translocate to the nucleus. There, they dimerize with HIF-1β/ARNT and bind hypoxia response elements (HREs) within the promoters of target genes. Together with the co-activator proteins p300 and CBP, the HIF complex activates the transcription of a panel of genes required for the response to hypoxia. OH = hydroxylation; Ub = ubiquitin.
Although low O2 is the principal regulator of HIF activity, it is clear that other signals influence HIF-α stability and function. Ionizing radiation, environmental stress, and angiopoietin-2 induce HIF-1α stabilization through the production of reactive oxygen species (ROS). ROS may reduce Fe2+ availability, thereby inhibiting PHD and FIH activity, resulting in HIF-α accumulation. 32 HIF-α levels can also be modulated by micro-RNAs, the small noncoding RNAs that regulate posttranscriptional stability and translation of target mRNAs. For example, miR-424 is induced by hypoxia in human umbilical vein endothelial cells (HUVECs) and promotes HIF-1α stability by inhibiting expression of Cullin-2, a scaffolding protein essential for assembly of the HIF E3 ubiquitin ligase complex. 33 In summary, HIF transcriptional activity is tightly regulated by multiple mechanisms and pathways, which suggests that precise control of its gene targets is necessary for tissue homeostasis and effective adaptive responses to low O2.
The HIF pathway mediates the primary cellular responses to low O2, which promotes both short- and long-term adaptation to hypoxia (Table 1). HIF rapidly increases O2 supply through upregulation of the vasodilatory enzyme inducible nitric oxide synthase (iNOS). 34 Nitric oxide (NO), the enzymatic product of iNOS, relaxes vascular smooth muscle cells, providing a short-term increase in blood flow. O2 demand is also lowered by increased utilization of glycolysis via induction of glycolytic enzymes, including phosphoglycerate kinase and Aldolase A, glucose uptake through increased glucose transporter-1 (GLUT-1) expression, and inhibition of mitochondrial respiration by upregulation of pyruvate dehydrogenase kinase (PDK1).35-37 O2 demand is further limited through decreased cell proliferation via HIF-mediated upregulation of the cyclin-dependent kinase inhibitors p21 and p27. 38 Long-term adaptation is achieved primarily through relief of local hypoxia by stimulating angiogenesis. The HIF pathway regulates a host of pro-angiogenic genes (Table 2), including vascular endothelial growth factor (VEGF), angiopoietin-1, angiopoietin-2, Tie2, platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), and monocyte chemoattractant protein–1 (MCP-1) (Table 2). Indeed, ectopic stimulation of the HIF pathway is sufficient to induce localized angiogenesis in the absence of additional factors in a variety of tissues and contexts. 39 HIF-regulated pro-angiogenic factors execute the HIF-specific angiogenic program by increasing vascular permeability, endothelial cell proliferation, sprouting, migration, adhesion, and tube formation (Figure 3). The breadth of pro-angiogenic HIF target genes and the comprehensive list of angiogenic processes regulated by them truly make HIF a “master regulator” of angiogenesis.
Hypoxia-Inducible Factor (HIF) Transcriptionally Induced Genes

ALDA = aldolase A; ALDC = aldolase C; AMF = autocrine motility factor; Bcl-2 = B-cell leukemia/ lymphoma 2; BNIP3 = Bcl-2 nineteen kilodalton interacting protein 3; CATHD = cathepsin D; CCD1 = coiled-coil-DIX1; CXCR4 = CXC chemokine receptor 4; DEC1/2 = differentiated embryo-chondrocyte expressed gene 1/2; GLUT-1/3 = glucose transporter1/3; GAPDH = glyceraldehyde-3-P-dehydrogenase; HSP70 = heat shock protein 70; IGF2 = insulin-like growth factor 2; IGF-BP1/2/3 = IGF factor binding protein 1/2/3; KRT14/18/19 = keratin 14/18/19; LDHA = lactate dehydrogenase A; LRP1 = LDL receptor–related protein 1; Lox = lysyl oxydase; MDR1 = multidrug resistance 1; MIC2 = microneme protein 2; MMP2 = matrix metalloproteinase 2; NFκB = nuclear factor kappa B; NOS2 = nitric oxide synthase 2; NUR77 = nuclear receptor 77; PDK = pyruvate dehydrogenase kinase; PFK = phosphofructokinase; PGK = phosphoglycerate kinase; PKM = pyruvate kinase M; REDD1 = regulated in development and DNA damage responses 1; Ref-1 = redox factor–1; TGF-α = transforming growth factor–α; TGF-β = transforming growth factor–β; TPI = triosephosphate isomerase; UPAR = urokinase plasminogen activator receptor.
Pro- and Anti-Angiogenic Factors Induced by Hypoxia/Hypoxia-Inducible Factors

ADM = adrenomedullin; Ang-1/2 = angiopoietin-1/2; COX-2 = cyclo-oxygenase 2; DLL = delta-like ligand; FGF = fibroblast growth factor; Flt-1 = fms-related tyrosine kinase 1; Kdr = kinase insert domain containing receptor; MMP = matrix metalloproteinases; NOS = nitric oxide synthases; PAI-1 = plasminogen activator inhibitor–1; PLGF = placenta growth factor; PDGF-B = platelet-derived growth factor beta; SCF = stem cell factor; SDF-1 = stromal-derived growth factor; Tie-2 = TEK tyrosine kinase endothelial; TIMP = tissue inhibitor of metalloproteinases; VEGF = vascular endothelial growth factor; VEGF-R = VEGF receptor.
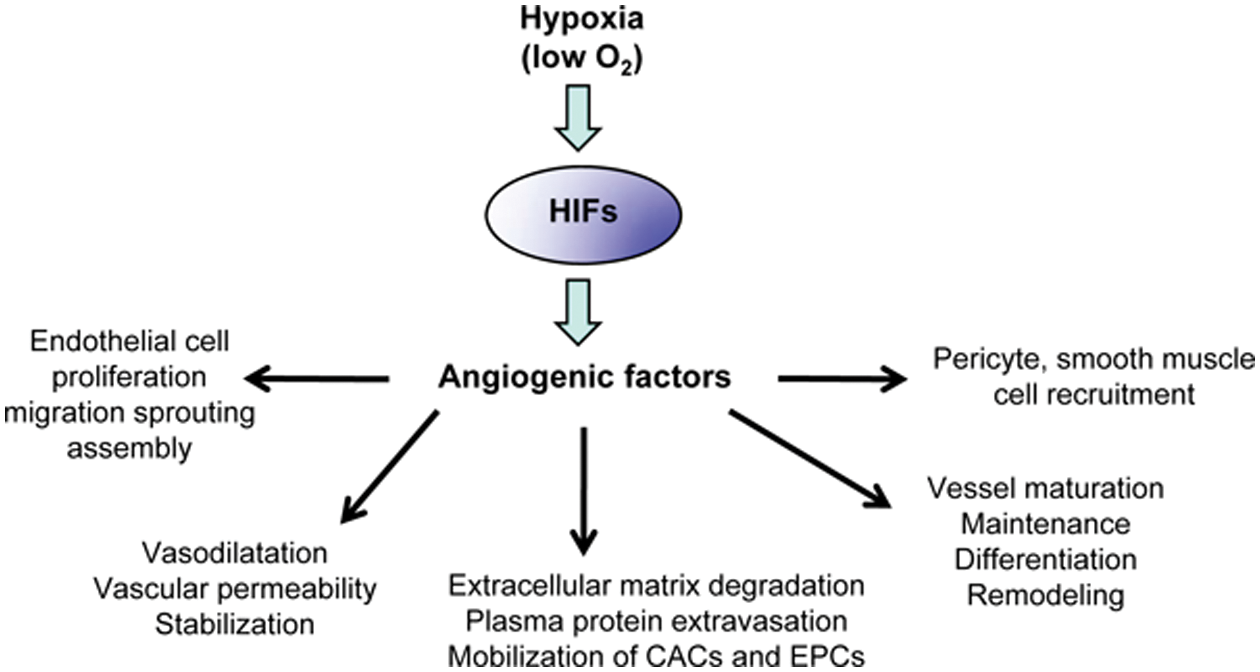
Regulation of angiogenesis and angiogenic steps by oxygen availability through the hypoxia-inducible factor (HIF)–induced angiogenic factors. It is now well established that, in response to hypoxia, HIFs regulate angiogenic genes (see Table 2) to control angiogenic steps and functions in embryonic vascular development and pathologic settings such as cancer or vascular diseases. CACs = circulating angiogenic cells; EPCs = endothelial progenitor cells.
Hypoxia and HIF Signaling in Physiological and Pathophysiological Angiogenesis
Hypoxia, the HIFs, and Vascular Development
Embryonic development presents a complex, dynamic environment for cells. Rapidly expanding tissues must coordinate growth with O2 delivery, as failure to do so deprives nascent organs of energy and nutrients. O2 acts as a molecular signal and, through its availability, coordinates blood vessel growth with the metabolic demands of growing tissues. Further complicating this scenario, mammalian development occurs in the relatively O2 poor uterine environment, with O2 concentrations ≤3%. In early embryogenesis, oxygenation of a cell aggregate can be maintained by simple O2 diffusion. However, once the diffusion limit of O2 is reached, a dedicated O2 delivery system must be developed to meet the ever-increasing demand for oxygen. The primitive vascular system is formed by a process called vasculogenesis, where progenitor cells form an immature vascular plexus de novo. Angioblasts, migratory vascular progenitor cells, proliferate and coalesce into cords to form the vascular rudiments. Development of the vascular system is coupled to primitive yolk sac hematopoiesis by hemangioblasts, which give rise to both angioblasts and hematopoietic stem cells. Hemangioblasts derived from the extraembryonic mesoderm (yolk sac) display this developmental potential, whereas those from intraembryonic (splanchnopleural) mesoderm generate definitive hematopoietic cells in the dorsal aorta through a hemogenic endothelium intermediate.40,41 In vitro, hypoxia promotes hemangioblast specification from mesodermal progenitors in an Arnt-dependent manner. 42 Low O2 is also known to influence differentiation of endothelial progenitor cells (EPCs), promoting an arterial fate over a venous one. HIF-1α regulates Hey2 and Delta-like 4 (Dll4) expression, both of which inhibit the venous-specification factor CoupTFII. 43 These studies suggest that hypoxia, via the HIF pathway, coordinates the earliest steps of vascular development.
After the primary vascular plexus is formed, endothelial cells (ECs) form new capillaries primarily by sprouting angiogenesis. A crucial mode of vessel formation during both embryonic development and adult life, sprouting angiogenesis forms new vessels by the outgrowth of ECs from existing vessels. As ECs are normally quiescent, a stimulating cytokine, such as VEGF, is required to commence angiogenesis. VEGF, considered a master regulator of angiogenesis in its own right, causes endothelial cells to detach from the parent vessel and migrate into the neighboring stroma. Hypoxia is the principal regulator of VEGF expression, as it is a direct transcriptional target of both HIF-1α and HIF-2α.44-48 Indeed, genetic ablation of Arnt results in embryonic lethality on embryonic day 10.5 due to defective vessel formation in the placenta, yolk sac, and branchial arches. 49 Arnt mutant vascular defects are accompanied by decreased VEGF expression, which is responsible for many of the vascular anomalies observed, as exogenous VEGF rescues these defects. Similarly, HIF-1α-deficient embryos die at E11 owing to similar vascular pathology, demonstrating the critical role of hypoxia and HIF signaling in cardiovascular development. 36 Although HIF-1α and HIF-2α are highly structurally similar and share many transcriptional targets (Figure 1), it is clear that they have divergent functions due to unique patterns of expression and transcriptional activity.13,50 For instance, Hif-1α is ubiquitously expressed, whereas Hif-2α expression is restricted to endothelial cells, type II pneumocytes of the lung, cardiomyocytes, macrophages, astrocytes, and the organ of Zukerlandl. 13 Although the Hif-2α knockout phenotype varies widely between genetic backgrounds, it is striking that the common feature to all the mutants is defective cardiovascular/pulmonary function. For instance, in one mouse strain, loss of Hif-2α causes improper vessel remodeling in the yolk sac and embryo, resulting in swollen and hemorrhagic vessels. 51 In another, Hif-2α ablation causes abnormal lung maturation due to defects in VEGF expression in the absence of apparent vascular deficits. 47 Finally, a third study demonstrated embryonic lethality due to bradycardia as the result of defective catecholamine synthesis. 52 Strategies to avoid the developmental defects from HIF-2α loss expand the role for HIF-2α in O2 delivery. Ubiquitous, acute postnatal deletion of Hif-2α results in anemia as a consequence of impaired EPO expression. 53 Furthermore, conditional deletion of Hif-2α in ECs reveals that HIF-2α promotes vessel maturation, as Hif-2α conditional knockout (KO) mice display vascular leakage in the lung and fat.54,55 These phenotypes are likely due to HIF-2α-mediated expression of genes involved in adhesion (integrins α9 and β2) and basement membrane formation (fibronectin). Interestingly, conditional HIF-1α deletion in ECs yields no developmental phenotype, although tumor angiogenesis is perturbed. 56 In contrast, endothelial deletion of Arnt results in embryonic lethality due to defective vascular development of the liver. 57 These observations highlight some of the overlapping and unique roles of HIF-1α and HIF-2α in vascular biology. In addition, these studies demonstrate the essential role of hypoxia signaling in embryonic vascular development, a process that also relies on the process of sprouting angiogenesis.
Hypoxia regulates sprouting angiogenesis at every step of this process through multiple pathways, including VEGF. First, activated migrating ECs must degrade the extracellular matrix (ECM), which is achieved by upregulation of matrix metalloproteinases (MMPs). MMP-2 is a direct transcriptional target of HIF-1α and mediates the migration of ECs in response to hypoxia. 58 In the extracellular matrix, newly formed ECs adhere to one another and form a network of trailing cells (“stalk” cells) and leading cells called “tip” cells. Gradients of chemotactic signals activate transmembrane receptors (e.g., VEGF-R2) on tip cells, stimulating their directed migration. Tip cell specification is negatively regulated by Notch signaling, which modulates vascular branching and morphogenesis through this mechanism.59-61 Interestingly, hypoxia may regulate vessel branching through modulation of Notch signaling. HIF-1α directly binds to the Notch intracellular domain (NICD) and augments its transcriptional activity. 62 Additionally, the Notch ligand Dll4 is a transcriptional target of both HIF-1α and HIF-2α in the endothelium.55,63 ECs trailing the leading edge begin to form tubes that extend the existing vascular network. Hypoxia enhances formation of endothelial tubes in vitro, and this pro-angiogenic effect is dependent on EC expression of HIF-1α. 56 Once established, the immature vasculature recruits vascular support cells, including pericytes and smooth muscle cells, and forms a basement membrane. This process, known as vessel normalization, involves HIF-2α, which directly regulates expression of the basement membrane component fibronectin. 55 These studies demonstrate that HIF regulates nearly every aspect of angiogenesis, which has made it an attractive therapeutic target in many human diseases.
Hypoxia, the HIFs, and Tumor Angiogenesis
In the early phase of malignant progression, a tumor may reside in a dormant state in an avascular area where the rate of cell proliferation is balanced with that of cell apoptosis. Subsequently, the tumor undergoes the so-called angiogenic switch, a transition from a nonangiogenic state to one of active vessel growth. 64 Angiogenesis is critical for tumor progression as tumor cell growth frequently outstrips the supply of O2 and nutrients. Indeed, inhibition of angiogenesis early in tumorigenesis impairs tumor progression in several inducible tumor models, including mouse models of islet cell and squamous cell carcinoma. 65 Conversely, depletion of the endogenous angiogenic inhibitors, thrombospondin-1 and thrombospondin-2, accelerates tumorigenesis in the preclinical model of spontaneous colorectal cancer (APCmin mice) and chemically induced skin carcinogenesis.66,67 Thus, angiogenesis is critical for both tumor formation and progression.
Despite active angiogenesis, tumor vessels are highly irregular, leaky, and poorly functioning. This characteristic leads to hypoxic domains and HIF-α stabilization, even in highly vascular tumors. Furthermore, factors associated with malignant transformation can stabilize HIF-α independently of hypoxia, such as Ras pathway hyperactivation, p53 mutation, and succinate accumulation.68-70 Hypoxia and HIF activation have profound effects on tumor biology, as HIF-1α and HIF-2α expression are associated with poor prognosis and metastatic disease in numerous cancers, including brain, breast, colon, head and neck, liver, lung, skin, and pancreas. 3 This is in part due to therapeutic resistance, as hypoxic tumors are refractory to radiation therapy because molecular O2 is required for the cytotoxic effects of ionizing radiation. 71 Chemotherapy is also ineffective in hypoxic tumors due to poor drug delivery and the HIF-mediated expression of ATP-dependent drug efflux pumps.72-74
Hypoxia and HIF pathway activation in tumor cells are an important stimulus for blood vessel growth. HIF-1α and HIF-2α regulate the expression of a suite of pro-angiogenic genes, including Vegf, Ang-1, Ang-2, and Tie-2, many of which themselves are used as biomarkers for tumor hypoxia (Tables 2, 3 and Figure 3). Indeed, HIF-1α directly regulates VEGF expression in vivo in Hepa-1 xenograft tumors. Hepa-1 cells with defective Arnt gave rise to poorly vascularized tumors with reduced VEGF expression. 75 VEGF expression is considered the primary effector for the angiogenic properties of HIF, whereas other transcriptional targets are clearly important for the full effect of HIF on vascular biology. For instance, Ang-2 is a direct HIF-2α target in ECs and may be indirectly regulated by HIF-1α, as the HIF-1α target VEGF can stimulate Ang-2 expression.55,76-78 Tie-2 receptor is antagonized by Ang-2, which regulates vascular remodeling in part by destabilizing existing vessels.79,80 Inhibition of Ang-2 with a neutralizing monoclonal antibody inhibited tumor angiogenesis, growth, and progression in mouse models, suggesting it plays an important role in tumor vascular biology. 81 A Tie-2 receptor agonist, Ang-1, is also induced by hypoxia in endothelial cells and pericytes. 55 Ang-1 promotes tumor angiogenesis by recruiting pericytes to maturing vessels. 82 Overexpressed in breast cancer 83 and glioma, 84 stem cell factor (SCF) is a HIF-1α transcriptional target that mediates neovascularization by enhancing EC survival, migration, 85 and EPC mobilization. 86
Main Angiogenic Molecules, Their Receptors, and Functions
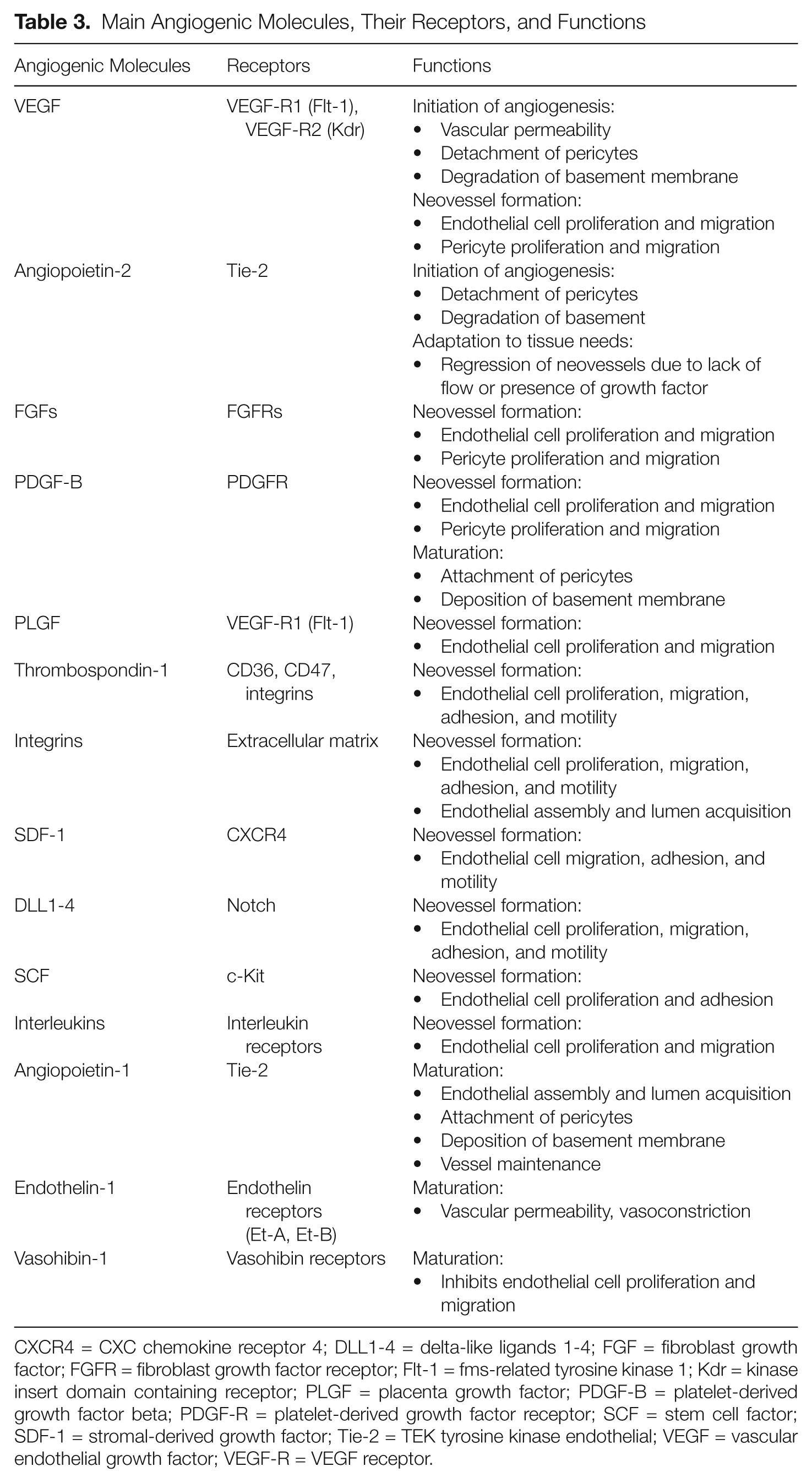
CXCR4 = CXC chemokine receptor 4; DLL1-4 = delta-like ligands 1-4; FGF = fibroblast growth factor; FGFR = fibroblast growth factor receptor; Flt-1 = fms-related tyrosine kinase 1; Kdr = kinase insert domain containing receptor; PLGF = placenta growth factor; PDGF-B = platelet-derived growth factor beta; PDGF-R = platelet-derived growth factor receptor; SCF = stem cell factor; SDF-1 = stromal-derived growth factor; Tie-2 = TEK tyrosine kinase endothelial; VEGF = vascular endothelial growth factor; VEGF-R = VEGF receptor.
In addition to the well-defined pro-angiogenic genes regulated by HIF, recent studies demonstrate that hypoxia can regulate additional effector molecules, which substantially extend the regulatory capacity of HIFs. Many microRNAs, which themselves are known to regulate multiple targets, are induced by hypoxia and the HIF pathway. In the endothelium, miR-210 is an HIF-1α target, and hypoxic miR-210 induction promotes endothelial tube formation and EC migration by downregulating Ephrin-A3. 87
In the endothelium, HIF-1α promotes expression of VEGF and its receptor VEGF-R2, thereby regulating an autocrine VEGF signaling loop that is vital to endothelial cell survival, proliferation, migration, and tube formation. 56 Accordingly, EC ablation of HIF-1α inhibits xenograft tumor growth due to reduced tumor angiogenesis. In contrast, xenograft tumors in mice with endothelial-specific Hif-2α deletion are smaller but have equivalent numbers of vessels. Hif-2α mutant tumor vessels exhibit defects in normalization, as Hif-2α mutant tumor vessels form but fail to luminize. 55 These studies demonstrate that in the tumor endothelium, HIF-1α and HIF-2α play complementary roles, where HIF-1α is essential for vessel growth, whereas HIF-2α promotes vascular maturation.
In addition to angiogenesis, tumor vascular development may also involve vasculogenesis, which is less well understood. The secreted ligand stromal-derived factor–1 (SDF-1) and its cognate receptor C-X-C chemokine receptor 4 (CXCR4) are thought to be the primary mechanism for the recruitment of bone marrow–derived cells to sites of vasculogenesis. 88 In fact, Sdf-1 is a transcriptional target of HIF-1α, and pharmacological inhibition of SDF-1/CXCR4 signaling decreased vascular regrowth after glioblastoma tumor irradiation.88,89 Intriguingly, inhibition of HIF-1α yielded a similar effect, collectively suggesting that hypoxia plays an important role in tumor vasculogenesis.
Several other bone marrow–derived cells (BMDCs), such as tumor-associated macrophages (TAMs), are also recruited to further accelerate active angiogenesis/vasculogenesis and promote invasion and metastasis. The HIF target genes Vegf, Fgfs, and Mcp-1 act as chemoattractive signals and recruit TAMs to hypoxic domains within tumors. Interestingly, TAMs themselves express HIF-1α and HIF-2α at high levels, and TAM HIF-2α expression is negatively correlated with positive outcomes in cervical cancer. 90 TAM HIF-2α expression promotes tumor progression, as macrophage-specific Hif-2α deletion results in diminished tumor progression in an inflammation-induced tumor model. 91 These results could be mechanistically explained due to changes in macrophage-modulated angiogenesis, as HIF-2α regulates CXCR4 and VEGF expression in macrophages. HIF-1α may also play a role in TAM-mediated angiogenesis, as HIF-1α regulates the glycolytic phenotype of TAMs and promotes migration, invasion, and aggregation of TAMs at sites of inflammation. 92 Indeed, HIF signaling regulates tumor vascular growth not only by regulating genes in tumor cells and the endothelium but also through recruitment of BMDCs.
HIF Inhibition as a Novel Cancer Therapy
Judah Folkman proposed targeted inhibition of the vasculature as a paradigm for cancer therapy almost 40 years ago. “Starving” a tumor of essential O2 and nutrients was shown effective in numerous preclinical trials and led to Food and Drug Administration (FDA) approval of a VEGF inhibitor (bevacizumab) for the treatment of metastatic colon cancer, metastatic breast cancer, glioblastoma, non–small cell lung cancer, and renal cell carcinoma. However, recent studies suggest single-agent anti-angiogenic therapy may actually promote tumor metastasis. 93 In mouse models of glioblastoma and pancreatic neuroendocrine cancer, administration of VEGF inhibitors (sunitinib, bevacizumab) or genetic deletion of Vegf in tumor cells showed potent antitumor activity. Surprisingly, treated mice experienced significant increases in localized tumor cell invasion and distant metastasis as a consequence of a VEGF blockade. A second study observed similar results in mouse xenograft models of breast cancer and melanoma. 94 Intratumoral hypoxia was drastically increased as a result of impaired angiogenesis. 93 As hypoxia is well known to promote migratory and invasive behavior of tumor cells, it is likely that induction of the HIF pathway and its well-characterized transcriptional targets (e.g., CXCR4, MMP-2, c-MET; Table 1) underlies this phenotype. Thus, the effects of tumor hypoxia resulting from anti-angiogenic therapies should be considered in cancer therapeutics.
Tumors often evade single-agent anti-angiogenic therapies through the selective activation of alternate pro-angiogenic pathways. 95 Consequently, simultaneous blockade of multiple pro-angiogenic pathways is more effective than single-agent anti-angiogenics. 96 Although it is unclear whether blocking multiple pathways will prevent the metastatic phenotype, sunitinib, a broad receptor tyrosine kinase inhibitor that blocks both VEGFR2 and PDGFR, causes a metastatic phenotype in mouse models. 93 Nonetheless, it will be important to determine if more effective anti-angiogenic agents also yield a metastatic phenotype. Since the HIF pathway regulates multiple pro-angiogenic pathways and promotes metastasis and invasion that may result from defective tumor angiogenesis, it is a highly attractive target for new cancer therapies. Recent studies have screened novel compounds and existing FDA-approved drugs for anti-HIF activity and found several new classes of drugs that may be useful for cancer treatments (Figure 4). For example, 2-methoxyestradiol97-99 and the cardiac glycosides100,101 digoxin, oubain, and proscillaridin A inhibit synthesis of both HIF-1α and HIF-2α. The chemotherapeutics anthracycline102,103 and doxorubicin 103 and the antibiotic echinomycin 104 attenuate HIF activity by inhibiting HIF-DNA binding, whereas acriflavine 105 interferes with HIF dimerization. Bortezomib 106 inhibits HIF-1α transactivation domain activity, whereas YC-1107,108 appears to function through an unknown mechanism. Importantly, cardiac glycosides, anthracyclines, and acriflavine markedly reduce tumor vascularization and tumor growth in an HIF-dependent manner. Defects in tumor angiogenesis were accompanied by reduced mobilization of CXCR4+/Sca1+, VEGFR2+/CD34+, and VEGFR2+/c-Kit+ circulating angiogenic cells (CACs) in a mouse xenograft model of prostate cancer.103,105 Since cardiac glycosides and chemotherapeutic drugs are already applied in clinical settings, their impact on HIF signaling has important implications for their use as anti-angiogenesis agents in cancer patients. However, none of the HIF inhibitors identified so far is specific to the HIF pathway, and several have significant side effects and toxicities that may preclude their use in the clinic. Therefore, development of HIF-specific inhibitors and validation of the clinical utility of nonspecific HIF inhibitors in anti-angiogenic cancer therapy represent a promising avenue for new cancer therapeutics.
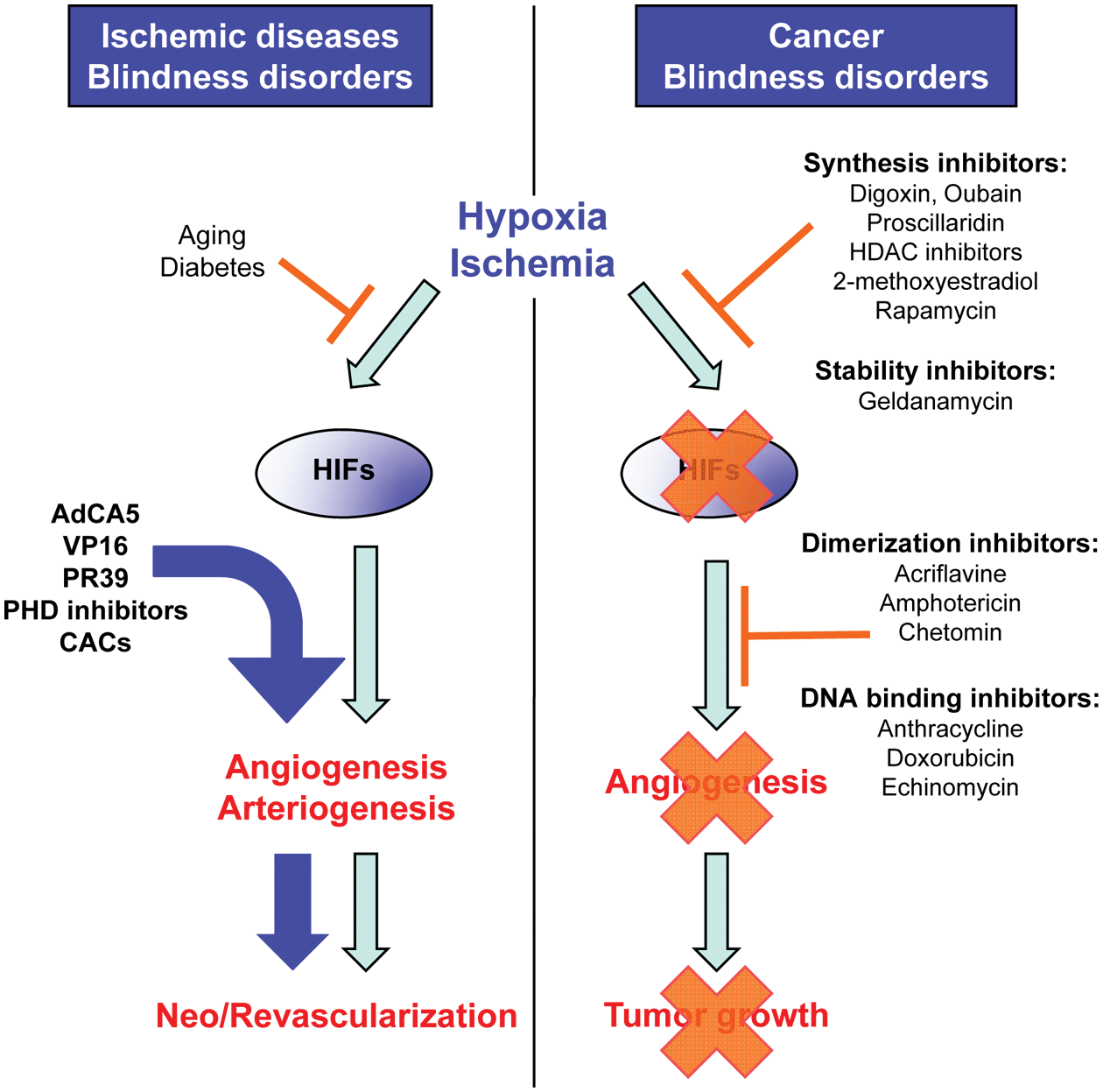
Determining the complex effect of local hypoxia on pathological angiogenesis is likely to have important therapeutic consequences. Since downregulation of the hypoxia-inducible factors (HIFs) inhibits angiogenesis and decreases tumor growth, regulation of the expression and activity of HIFs is evolving as a potential target for modulating tumor angiogenesis and is becoming an attractive approach for the treatment of solid tumors and macular degeneration, maladies typically associated with unregulated or excessive angiogenic activity. Inhibitors of HIF synthesis, stability, dimerization, and DNA binding are currently in clinical trials and show promising results. In contrast, forced expression or activation of HIFs, using viral vectors or prolyl hydroxylase (PHD) inhibitors, could evolve as a potential solution for the improvement of therapeutic angiogenesis and revascularization after critical ischemia in adults affected by ischemic diseases. The ability to induce and regulate angiogenesis and vascular remodeling in a directed manner would represent a major advance in the treatment of ischemic vascular diseases, including myocardial infarction, atherosclerosis, and peripheral artery disease. HDAC = histone deacetylase; CAC = circulating angiogenic cell.
Hypoxia, the HIFs, Angiogenesis, and Vascular Diseases
Occlusive vascular diseases remain the most important causes of death and morbidity in industrialized societies. 109 Treatment of end stages such as myocardial infarction, peripheral artery disease (PAD), and stroke is usually limited only to palliative interventions, such as angioplasty and, in severe PAD cases, limb amputation.110,111 The ability to induce and regulate angiogenesis and vascular remodeling in a directed manner would represent a major advance in the treatment of ischemic vascular diseases. Recent studies have demonstrated how hypoxia and the HIF pathway, through the modulation of angiogenic genes, such as Vegf (Table 3 and Figure 3), regulate the adult vascular system in numerous pathological conditions.88,112-116 Not surprisingly, the HIF pathway represents an attractive therapeutic target in ischemic diseases, and its continued study in these contexts has revealed important insights (Figure 4).
Hypoxia, the HIFs, and Peripheral Artery Disease
Peripheral artery disease is a condition characterized by obstruction of large arteries leading to vascular dysfunction in the extremities.110,117 Vessel occlusion in PAD is usually caused by the development of atherosclerotic plaques, which are initiated by injuries to the endothelium from hemodynamic stress and uptake of oxidized low-density lipoprotein. Endothelial damage stimulates rapid proliferation of vascular smooth muscle cells, thickening the arterial wall. Nascent plaques are subsequently infiltrated by large numbers of macrophages, which drastically increase metabolic demand and cause local tissue hypoxia and induction of an angiogenic response. 118 Plaque perfusion promotes expansion by allowing more efficient infiltration of macrophages through new capillaries. 119 Interestingly, HIF-1α and VEGF are expressed in the plaque, suggesting that the HIF signaling pathway is directly involved in plaque angiogenesis. 120 Continued plaque growth progressively narrows existing arteries, restricting blood flow and reducing perfusion to adjacent tissues. Atherosclerotic stenosis (narrowing) of peripheral arteries leads to critical limb ischemia in 1% to 2% of patients. 121 The most severe form of PAD, critical limb ischemia, is characterized by pain at rest, ulceration, and/or gangrene. Although a neovascular response in the affected limb is mounted, it is often insufficient to adequately reperfuse the ischemic tissue, which eventually requires amputation. The HIFs promote the neoangiogenic response to tissue ischemia and pathophysiological angiogenesis in PAD, as levels of HIF-α mRNA, protein, and transcriptional targets increase following ischemic insult in animal models.114,116 HIF transcriptional activity leads to the production of new capillaries via angiogenesis and the remodeling of existing arteries to accept increased flow, a process called arteriogenesis. 122 Surgical ligation of the femoral artery leads to a severe decrease in hindlimb blood flow, usually to <10% to 20% of the nonligated side, and serves as an animal model of PAD.123-125 Reduced blood flow in the main limb artery induces ischemia distally, which triggers a prototypical angiogenic and arteriogenic response. 126 The HIFs play an important role in the blood flow recovery during hindlimb ischemia, which is mainly achieved by arteriogenesis while angiogenesis plays a secondary role.127,128 For example, femoral artery ligation (FAL) experiments revealed decreased limb perfusion and increased spontaneous amputation in Hif-1α +/– mice, which have a severely blunted induction of HIF-1α protein in response to hypoxia. 116 Administration of the HIF-1α inhibitor 2-methoxyestradiol phenocopied the defects observed in Hif-1α +/– mice. In contrast, forced expression of a constitutively active form of HIF-1α that is resistant to O2-dependent regulation, AdCA5, 112 stimulated reperfusion following FAL in both mouse and rabbit models.114,129 AdCA5 treatment significantly increases the arteriogenic response, characterized by enlarged collateral blood vessels, demonstrating that HIF activity promotes vessel remodeling in addition to angiogenesis.
Recent work suggests several risk factors for PAD may mechanistically operate through the HIF pathway. As mice age, the ischemic accumulation of HIF-1α and its transcriptional targets diminishes following FAL. 116 Consequently, recovery from reperfusion is also impaired in old mice, which exhibit increased frequency of spontaneous limb amputation. Reduced HIF target gene expression in the limbs of old mice correlates with observed deficits in reperfusion. Diabetic mice also exhibit defective reperfusion following FAL and impaired wound healing. 130 When exposed to high glucose and low O2, dermal fibroblasts derived from Leprdb/db diabetic mice fail to stabilize HIF-1α protein and induce VEGF.131,132 Expression of HIF-1α and VEGF levels is attenuated in diabetic mice subjected to FAL, suggesting defective HIF induction may underlie some of the vascular complications common to diabetics.133,134 Mechanistically, diabetic mice exhibit defects in HIF signaling because high glucose impairs the activity of HIF-1α by inhibiting HIF-1α/Arnt heterodimerization and HIF-1α/p300 binding. Therapeutic enhancement of HIF activity can overcome age and diabetes, as ectopic expression of HIF-1α can partially rescue limb perfusion in old mice, 116 whereas activation of the HIF pathway via PHD inhibition reverses deficits in neovascularization observed in diabetic mice. 135 The importance of aging and diabetes as modulators of ischemic responses is becoming increasingly clear and will likely have significant functional importance for all ischemic diseases.
In addition to angiogenesis and arteriogenesis, the mobilization of circulating CACs appears to play an important role in the vascular response to ischemia. CACs are a heterogeneous cell population derived from the bone marrow that includes endothelial progenitor cells, circulating endothelial cells, hematopoietic progenitor cells, and mesenchymal stem cells.136-138 Stimulating cytokines such as HIF targets VEGF, placenta growth factor (PLGF), and SDF-1 induce CAC migration from the bone marrow and into the circulatory system. 139 CACs migrate to sites of ischemia where they promote vascular remodeling and stimulate angiogenesis and arteriogenesis. In models of ischemia-induced neovascularization, including FAL, tumors, wound healing, and choroidal neovascularization, CACs have been shown to incorporate into new blood vessels and promote neovascularization.116,129,134,137,140-145 SDF-1/CXCR4 and VEGF/VEGF-R2 signaling regulate the recruitment of these cells. 146 Ectopic expression of HIF-1α enhances the mobilization and recruitment of CACs to ischemic sites in the FAL model by upregulating SDF-1 and VEGF. 116 Consistently, diabetic mice also exhibit reduced CAC activity, which can be rescued by forced expression of HIF-1α or PHD inhibition via desferrioxamine (DFO) or dimethyloxalyglycine (DMOG).133,135,147 HIF-1α has been heavily scrutinized in preclinical studies of PAD, but the role of HIF-2α is less clear.
Given that HIF-2α is highly expressed in the endothelium, it is likely to play a role in PAD. Indeed, studies in Phd1 knockout mice provide further evidence that HIF-2α can buffer tissues from hypoxic/ischemic stress.148,149 When placed under ischemic stress, Phd1–/– limb skeletal muscle is protected from oxidative damage and cell death. The ischemic tolerance in Phd1–/– skeletal muscle is predominantly dependent on HIF-2α, as simultaneous deletion of both Phd1 and Hif-2α blocked the protective effects of PHD1 deficiency. HIF-2α may promote this tolerance by modulating glucose metabolism indirectly through regulation of PPARα, which is essential for ischemic tolerance in Phd1-deficient skeletal muscle. 148 PDK4, which limits mitochondrial glucose metabolism by inhibiting the pyruvate dehydrogenase complex, is a target of PPARα. 150 Alternatively, HIF-2α may contribute to the ischemic phenotypes in Phd1–/– skeletal muscle by regulating redox homeostasis.151,152 These studies demonstrate the diverse mechanisms through which hypoxia and the HIF pathway regulate vascular responses to tissue hypoxia and ischemia.
Hypoxia, the HIFs, and Ischemia-Induced Coronary Collateralization
In addition to its role in PAD, atherosclerosis also affects the coronary arteries, where atherosclerotic stenosis and luminal occlusion induce myocardial infarction and ischemia/hypoxia stimulates collateralization. Sixty percent of patients with coronary artery disease develop collateral vessels that bypass the stenosis.153,154 The presence of collaterals is associated with reduced infarct size, less severe functional deterioration, and reduced mortality following myocardial infarction.110,117,153,154 Patients who do not develop collateral vessels to increase blood flow distal to the site of stenosis are more likely to develop heart failure. 154 Recent studies indicate that deficits in the hypoxic response underlie phenotypic heterogeneity observed with respect to collateralization. For instance, monocytes isolated from patients with coronary artery disease (CAD) with collaterals produce greater amounts of VEGF in response to hypoxia compared to monocytes from patients without collaterals. 155 Moreover, increased HIF-1α expression in other blood cells (e.g., leukocytes) has also been associated with the presence of coronary collaterals in patients with CAD. Following this observation, the frequency of single-nucleotide polymorphisms (SNPs) in the human HIF1A gene was significantly higher in CAD without collaterals and also increased in patients with stable exertional angina over those presenting with myocardial infarction.156,157 These HIF1A variants are less functional, as Hif-1α null mouse embryonic fibroblasts (MEFs) transfected with the HIF1A variants fail to upregulate HIF target genes as well as wild-type HIF1A. Defective HIF-1α activity, therefore, may lead to early onset symptoms. Genetic screening for less functional HIF-1α variants could aid early clinical evaluation of patients who are likely to develop advanced coronary disease.
Activation of HIFs in Ischemic Diseases as a Novel Therapy
Administration of angiogenic factors, including VEGF and FGF, and transplantation of bone marrow cells have been tested as potential treatments to enhance vascularization in ischemic diseases. However, increasing HIF-α levels and activity may represent a superior therapeutic approach due to the multiple pro-angiogenic pathways it regulates (Figure 4). Blood vessels formed in pathological conditions are typically abnormal, tortuous, and leaky. These types of blood vessels are often observed experimentally when induced with a single agent, such as VEGF. To best treat ischemic diseases, normal vessels need to be formed. Interestingly, mice expressing constitutively active forms of HIF-1α and HIF-2α that are refractory to O2-dependent regulation are hypervascular,158-160 but in contrast to VEGF overexpressing mice, they possess “normalized” vessels. 159 In a tumor vessel abnormalization model, endothelial PHD2, via regulation of HIF-2α, senses and readapts O2 supply in response to O2 deprivation. 161 Haplodeficiency of Phd2 in ECs does not affect tumor vessel density and area, tortuosity, or lumen size but induces “normalization” of the endothelial lining, barrier, and stability. Interestingly, tumor vessels in Phd2+/– mice are lined by a single-monolayer phalanx of regular, orderly formed, polarized cobblestone ECs, which have few fenestrations. These changes in EC shape, not numbers, do not affect primary tumor growth but improve tumor perfusion and oxygenation. 161 Inhibition of PHD2 in ECs would provide a conceptually different strategy to blocking angiogenesis, whereby tumor vessel function is improved by streamlining the EC layer and improving delivery of oxygen to tumors. This would render the tumors less malignant and metastatic, making them more vulnerable to radiation and chemotherapy.
Several strategies to promote HIF activity and angiogenesis are in development for use in ischemic diseases and have been employed in various preclinical models. Exposure to mild hypoxia, called hypoxic preconditioning, protects against ischemic challenge by inducing HIF-α accumulation. This approach can prevent apoptosis, which is seen in the retina where hypoxic preconditioning protects photoreceptors from light-induced damage. 162 Angiogenesis is also stimulated by hypoxic preconditioning, which has been documented in the myocardium. 163 The therapeutic utility of this intervention is limited, given that hypoxic treatment must precede the ischemic stress. Alternatively, ectopic expression of the HIF-α subunits represents an attractive option. Electroporation of plasmid DNA encoding HIF-1α enhanced the angiogenic response in models of hindlimb ischemia and wound healing.116,133,135,164 Consistent with this finding, adenoviral delivery of an O2-refractory form of HIF-1α exhibited benefit in limb ischemia models in aged and diabetic mice.116,129,165 A similar approach, treatment with DNA encoding the N-terminus of HIF-1α fused to the VP16 transactivation domain, also stimulated angiogenesis in animal models166-168 and is currently progressing through phase I and II clinical studies in patients with severe PAD. 169
These approaches, however, suffer from many of the challenges common to gene therapy and must be cautiously evaluated. An alternative approach to enhancing HIF signaling is to inhibit HIF-α degradation. Proteasomal inhibition with the macrophage-derived peptide PR39 inhibits HIF degradation and increases angiogenesis in ischemic mouse cardiac tissue. 170 As the PHD enzymes are the primary O2 sensing proteins, they have been pharmacologically targeted with the aim of augmenting HIF activity. As stated previously, PHD enzymes require O2, iron, and 2-oxoglutarate (2-OG) for their enzymatic activity. 16 Accordingly, iron chelators, such as DFO, exhibit potent HIF-α stabilizing activity both in vitro and in vivo.17,171 2-OG analogues can also inhibit PHD activity by displacing the 2-OG cofactor in the PHD enzyme. DMOG is a cell-permeable precursor to the 2-OG analogue N-oxalylglycine that is effective both in vitro and in vivo. 16 PHD inhibitors are not specific to PHD1-3, as they likely inhibit all iron and 2-OG-dependent dioxygenases, of which 60 to 80 have been identified in humans. For example, lysyl hydroxylase, which is important in collagen biosynthesis, would also be affected.172,173 Therefore, caution should be used when drawing conclusions based solely on PHD inhibitors, as the off-target effects of these compounds are not fully understood.
Hypoxia and HIFs in Blindness Disorders
Uncontrolled blood vessel growth is a central pathological component of many human blindness disorders, including diabetic retinopathy, age-related macular degeneration (AMD), glaucoma, and retinopathy of prematurity (ROP). Neuronal cell death and vision loss observed in these diseases are caused by aberrant, leaky vessels, which are often associated with pathological neovascularization. Collectively, these diseases are the most common cause of vision loss in America today, suggesting that highly effective anti-angiogenic therapy would have a tremendous effect on the prevalence of blindness in the industrialized world. 174 In fact, neovascular eye diseases are the clearest example of the therapeutic utility of blocking angiogenesis, as VEGF inhibitors have been successfully employed in the clinic and shown efficacy in treatment of the wet (neovascular) form of AMD. 175
Although strategies to inhibit VEGF have found success in the clinic, it is clear that other pro-angiogenic factors play an important role in this disease, as anti-VEGF therapies are ineffective in approximately 50% of patients.175,176 Indeed, simultaneous blockade of multiple pro-angiogenic factors yielded significantly better results in mouse models of ocular neovascularization. 96 Given that the HIF pathway is a master regulator of angiogenesis and modulates multiple pro-angiogenic pathways, it is an attractive target for new therapeutic strategies (Figure 3). Although the role of HIFs in neovascular eye disease has not been extensively evaluated, HIF-1α and HIF-2α are expressed in ECs and macrophages from neovascular membranes harvested from AMD patients. 177 Furthermore, HIF-1α and HIF-2α are both induced in a cell type–specific manner in a murine model of ROP, suggesting hypoxia plays a role in these diseases. 178 Indeed, the HIF pathway has a functional role in ROP, as systemic reduction of HIF-2α expression with a hypomorphic Hif-2α allele caused marked decreases in retinal neovascularization that was accompanied by defects in EPO expression. 179 Moreover, specific deletion of Hif-2α in astrocytes attenuated the neovascular response in the murine ROP model but did not affect developmental retinal angiogenesis. 180 The functional role of HIF-1α in pathological ocular neovascularization is less clear, although its integral role in other neovascular models strongly suggests it is an important regulator of retinal neovascularization. Indeed, the HIF inhibitor digoxin effectively suppressed neovascularization in mouse models of ROP and wet AMD. 181 Both models exhibited attenuated HIF-1α accumulation, VEGF induction, and recruitment of macrophages and bone marrow–derived cells, suggesting HIF inhibition blocks many processes associated with ocular neovascularization. Although off-target effects of digoxin cannot be completely ruled out, these data provide a valuable proof of concept that HIF inhibition can effectively treat neovascular eye disease.
Therapeutic enhancement of HIF activity may also represent an important preventive therapy for retinopathy of prematurity, the second most common cause of blindness for children younger than 6 years old. ROP is a neovascular eye disease that affects premature infants. The retinal vasculature forms late in embryonic development and is thought to be regulated by retinal hypoxia. As the embryonic environment is relatively hypoxic, premature infants experience relative hyperoxia, which inhibits vascular growth and obliterates newly formed vessels of the eye. Continued retinal development increases the metabolic demands of the now hypoperfused retina and generates areas of hypoxia that promote a strong neovascular response. Retinal detachment, hemorrhage, and retinal damage are caused by neovascularization in ROP, leading to severe vision loss and blindness in some cases. Given the pathogenesis of this disease, it is not surprising that hypoxia plays a critical role in ROP. Pharmacologic activation of the HIF pathway by administration of the PHD inhibitor, DMOG, during the hyperoxic vaso-obliterative phase of ROP significantly abrogates vessel destruction and the resultant neovascularization in a mouse model. 182 Thus, therapeutic blockade and enhancement of HIF activity represent viable strategies for the treatment of many human blindness disorders (Figure 4).
Concluding Remarks and Future Directions
Intensive studies have clearly established the hypoxia/HIF signaling pathway as a master regulator of the vascular system (Figure 3). Accordingly, it represents an important therapeutic target for vascular diseases and cancer. The ability to induce and regulate angiogenesis and vascular remodeling through targeted manipulation of the HIF pathway would be a major advance in the treatment of ischemic vascular diseases, including myocardial infarction, atherosclerosis, and PAD, which constitute a major cause of death and morbidity in the industrialized world. HIF therapy holds significant advantages over current treatments for ischemic vascular diseases, as it can generate new functional vessels to relieve ischemic stress (Figure 4). Moreover, strategies that focus on one or more downstream effectors of HIF, such as VEGF, produce leaky, aberrant vessels, whereas induction of HIF-1α or HIF-2α induces formation of normalized better functioning vessels. Therefore, induction of HIF activity is likely the best molecular target for therapeutic intervention in the vascular system.
Future studies will need to define the parameters for HIF therapy in various preclinical models. Given that HIF-1α and HIF-2α have partially redundant and unique transcriptional identities, it will be important to determine whether expression of one or both of these factors will yield optimal results. Along these lines, the role of HIF-2α in models of ischemic vascular disease is not well understood. Recent data demonstrate that HIF-2α promotes vessel remodeling and maturation, indicating that enhanced HIF-2α activity may improve therapeutic outcomes. The amount and duration of HIF activity will need to be examined to determine proper treatment paradigms. The threshold for the duration of HIF activity required for a pro-angiogenic response may be quite low, as recent experimental data show that even transient local expression of a constitutively active form of HIF-1α yielded a beneficial pro-angiogenic response in patients with limb ischemia. 169 Importantly, this strategy circumvents issues inherent to gene therapy approaches and alleviates concerns about toxicity or adverse effects associated with PHD inhibitors or systemic HIF activation.
Targeted inhibition of the HIF pathway with the intent of blocking angiogenesis is of great interest for the treatment of cancer and neovascular diseases of the eye (Figure 4). Tumors often evade single-agent anti-angiogenic therapy through induction of alternate pro-angiogenic pathways and increased metastasis.93,183 Moreover, anti-VEGF treatment is ineffective in nearly 50% of wet AMD patients, whereas blockade of multiple pro-angiogenic pathways simultaneously yields better vascular responses in mouse models of both cancer and neovascular eye disease.96,176 Since HIF regulates multiple pro-angiogenic pathways, targeted inhibition of the HIF pathway may provide increased therapeutic outcomes for those with neovascular eye disease. HIF pathway inhibition may prevent increased metastasis that some have reported in response to anti-angiogenic therapy, as it promotes increased migration and invasion of tumor cells in response to hypoxia. All of the HIF inhibitors described to date are nonspecific, and many have a very narrow therapeutic window due to significant toxicities. Therefore, development of HIF-specific inhibitors will be an important area of future investigation.
The importance and role of CACs and EPCs in ischemic vascular disease are still under debate, although there are compelling data that they promote vascular repair and vessel remodeling. Bone marrow–derived cells are clearly recruited to areas of hypoxia in an SDF-1 and CXCR4-dependent manner.139,146 Increased HIF-1α promotes recruitment of CACs and EPCs, whereas inhibition of HIF-1α decreases CAC and EPC recruitment following hindlimb ischemia. 116 The role of HIF-2α in this process is unknown, although HIF-2α preferentially regulates CXCR4 in tumor-associated macrophages, 91 suggesting it may play a role in this process. It will be interesting to evaluate if HIF-2α regulates EPC biology and determine its functional significance in models of ischemic neovascularization.
Although the current transcriptional repertoire of the HIFs is very broad, there is reason to suspect that HIF regulates the endothelium through additional mechanisms and pathways. For instance, recent studies demonstrated that Wnt/β-catenin signaling plays an important role in vascular development and disease through regulating processes that include endothelial cell differentiation, morphogenesis, and blood-brain barrier formation.184-188 Interestingly, hypoxia via the HIF pathway displays a context-dependent regulation of the Wnt pathway. In stem cells, HIF-1α promotes Wnt/β-catenin signaling, whereas in cancer cells, it inhibits Wnt signaling.189,190 It will be interesting to see how hypoxia and the HIF pathway regulate Wnt in this context and evaluate the functional significance of this signaling axis in vascular diseases.
Finally, although many of the molecular targets proposed for the inhibition of angiogenesis appear promising, particularly through targeting HIF and related pathways, caution should always be used when applying findings from in vitro studies to clinical applications. As with most studies using isolated systems and molecular targets, many of the results obtained in preclinical studies have yet to be verified as relevant in a clinical setting. Moreover, the significance of targeted HIF therapy is still unknown in a clinical setting, and many of the preclinical studies have found conflicting results, indicating that the biology of HIF is still not completely understood. Nonetheless, there is tremendous therapeutic potential for targeted manipulation of the HIF pathway in a variety of human diseases.
Footnotes
Acknowledgements
We thank the Simon laboratory for helpful discussions and comments. We thank Sarah Scotland, Erin H. Graf, and Dr. Vijay Dondeti for editing. We apologize to other authors of original reports not included in the references section because of space constraints.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was funded by the Howard Hughes Medical Institute, Abramson Family Cancer Research Institute, and the National Institutes of Health (HL66130). B.L.K. was supported by a NRSA fellowship from the NEI (EY021411).