
Abstract
The beginning of this century was not only marked by the publication of the first draft of the human genome but also set off a decade of intense research on epigenetic phenomena. Apart from DNA methylation, it became clear that many other factors including a wide range of histone modifications, different shades of chromatin accessibility, and a vast suite of noncoding RNAs comprise the epigenome. With the recent advances in sequencing technologies, it has now become possible to analyze many of these features in depth, allowing for the first time the establishment of complete epigenomic profiles for basically every cell type of interest. Here, we will discuss the recent advances that allow comprehensive epigenetic mapping, highlight several projects that set out to better understand the epigenome, and discuss the impact that epigenomic mapping can have on our understanding of both healthy and diseased cells.
What Is the Epigenome?
One of the enduring mysteries of biology is that we all start off as a single cell but that, instead of becoming an ever-expanding mass of identical cells, we are made up of a wide variety of specialized tissues, despite carrying basically the same DNA in each cell. Somehow, each of the 200 different kinds of cells in the human body must be reading off a different set of the hereditary instructions written into the DNA. 1 It has become clear that the availability of these hereditary instructions is mediated through a vast suite of epigenetic marks. These comprise a variety of molecular and structural modifications to DNA that do not change the underlying sequence but ensure that the right genes are expressed at the right time. 2 As such, each cell type, including not only healthy but also diseased cells, is thought to have a unique epigenetic state that determines its identity. 3 The epigenetic state of the cells represents the sum of developmental and physiological influences. As such, the epigenome provides a crucial interface between the environment and the genome. Indeed, various factors such as nutrition, toxins, drugs, infection, disease state, and exposure to toxic agents are known to affect DNA and histone modifications.4,5 Epigenetic states have been described that are meiotically heritable, whereas others can only be propagated through mitosis. 6 Other chromatin changes are even more transient and coupled solely to active gene transcription. Although all marks are part of the cellular epigenome, the extent to which an epigenetic mark is stable determines to what extent it can embody cellular memory.5,7
With the development of new techniques that allowed investigation of larger genomic regions or even an entire mammalian genome, epigenetic research has changed dramatically over the last decade, as exemplified by the vast increase in the number of publications mentioning the word “epigenome” over the last 10 years (Fig. 1). Initially, epigenetic research was focused on single loci combining, for example, chromatin immunoprecipitation with quantitative PCR analysis. This was followed by techniques based on combining chromatin immunoprecipitation (ChIP) or other chromatin enrichment methods with DNA hybridization to either custom or commercial tiled oligonucleotide microarrays (ChIP-chip). Although these techniques allowed for the first time global analysis of epigenetic marks, the sequence resolution and genomic coverage per microarray were relatively low. Moreover, as it required multiple arrays to cover a complete genome of higher eukaryotes, the technique was very costly in order to generate comprehensive views on chromatin modifications. However, nowadays, with the widespread adoption of next-generation sequencing (NGS), genome-wide epigenetic mapping experiments can be performed with unprecedented resolution,5,8-10 and this has truly opened the field of epigenomics. Indeed, many projects throughout the world have been initiated to shed more light on epigenetic phenomena at the genome-wide level. Here, we will discuss the different aspects that need to be taken into account when starting large-scale epigenetic projects and will also examine the current state of epigenomic research.
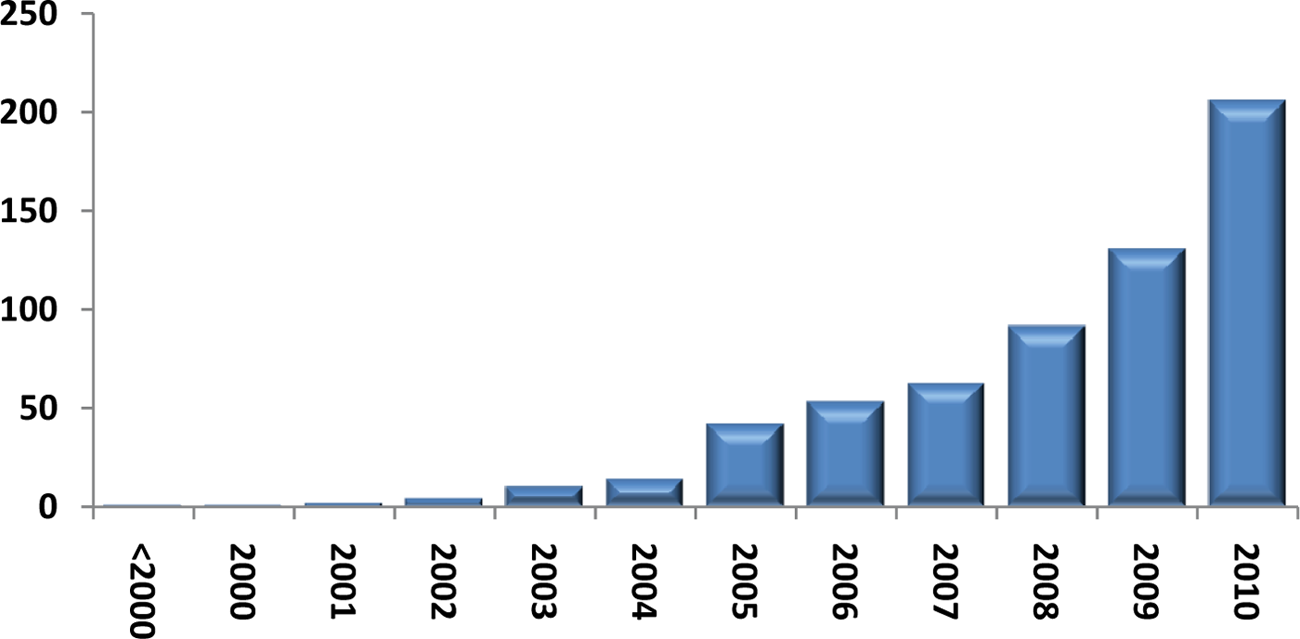
Scientific publications since 2000 mentioning the word “epigenome.”
What Is Worthwhile Mapping?
Several aspects need to be taken into account when expanding epigenetic research to the genome-wide level. The first is to determine what would be most interesting to map. As every researcher probably favors his or her own model system, the answer to this question will be similar as for the DNA sequencing projects: in the end, we would like to know the full epigenome of all organisms of potential interest. Indeed, with the rapid increase in sequence technology and following the explosion of fully sequenced organisms in the last decade, it can be expected that in the coming decade, mapping of epigenomes will become routine business, ultimately resulting in establishing the epigenomic maps of most human tissues and cell types as well as those of many model organisms. However, as current resources are still limited, choices have to be made in the selection of cell types and epigenetic features that can be included into epigenomic projects. 5 An important consideration is the amount of material that can be used. Because current technologies require substantial amounts of a rather homogenous cell population as a starting point, this is still one of the limiting steps for establishing complete epigenomic maps. Although model cell lines are ideal in terms of homogeneity, it has become clear that their epigenomic make-up is far from representative of the cells that constitute the tissue they are derived from. On the other hand, many human tissues are very heterogeneous and yield insufficient cells of a single type to permit comprehensive epigenetic analysis. As a result, current efforts mainly focus on cell populations that can be obtained in great quantity and that are fairly homogenous such as CD4+ cells.11,12 To bypass this problem, pooling samples would be an option, although this again is a less optimal solution as one has to consider the variation that is being introduced within the source material. However, it has to be noted that although current techniques require substantial amounts of material, these problems are expected to be overcome at least partially by the introduction of new techniques that require less starting material.13,14 In summary, each epigenomic project has to carefully seek a balance between the scientific priorities, the availability of relevant (tissue) materials, and the techniques that can be utilized, in addition, of course, to the budget that can be allocated.
How to Map the Epigenome?
Current technology allows for the first time the comprehensive mapping of all different aspects of the epigenome: ChIP-seq can be used to examine histone modifications at the genome-wide level, RNA-seq for profiling the transcriptome, and a variety of techniques are available to examine global accessibility and the DNA methylome (Fig. 2). An important consideration for epigenome analysis is the establishment of standard operating procedures (SOP) that allow data of the highest quality to be generated and compared through similar efforts among different consortia. Currently, IHEC, an international consortium that sets out to establish high-resolution reference epigenome maps for the research community, aims to define these standards, not only for data generation but also for data storage and availability. This should be the primary guideline for every effort into epigenome generation throughout the world.
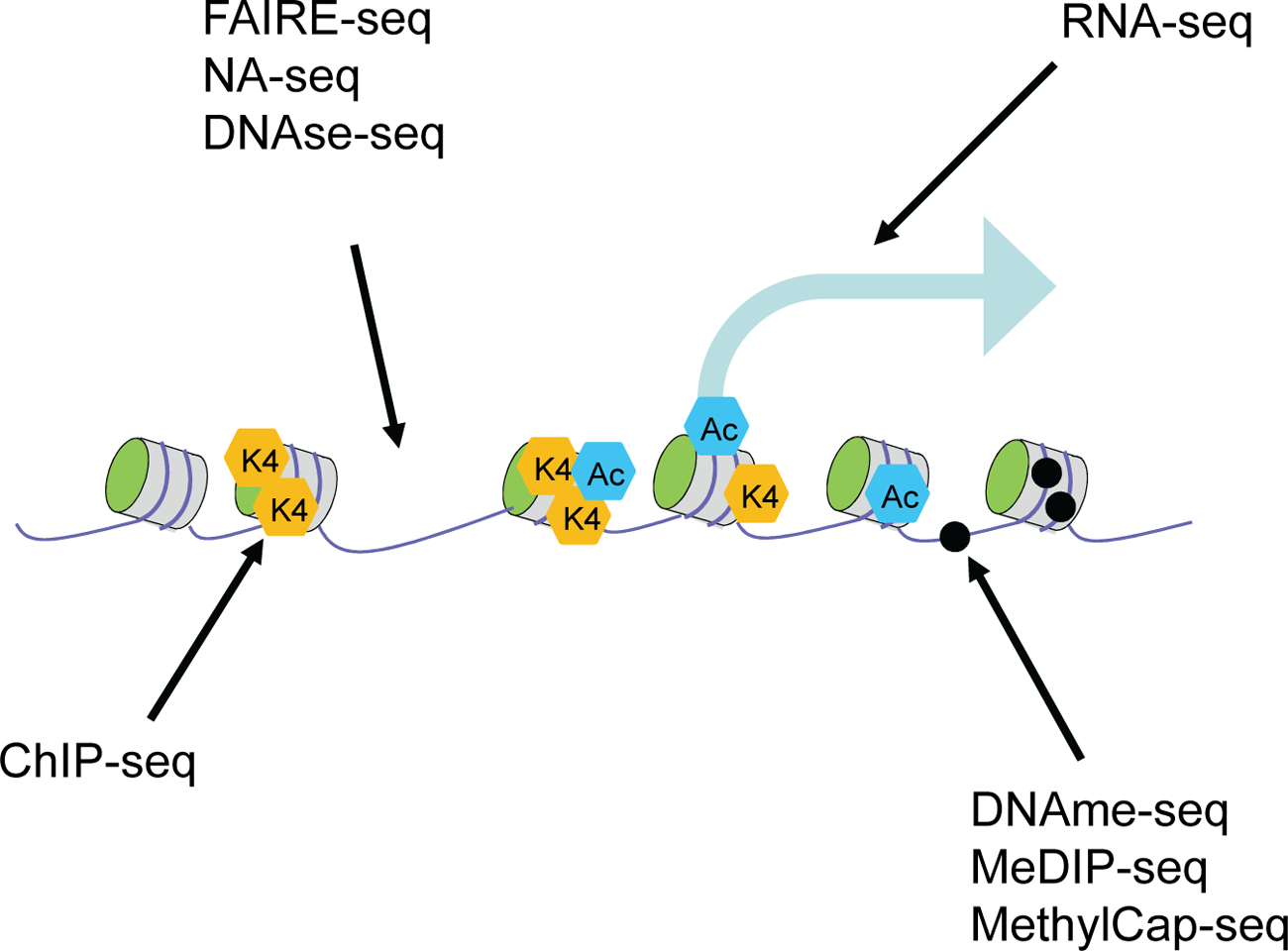
Current technologies used to map epigenomes.
ChIP-Seq
The aim of ChIP-seq experiments is to map all in vivo DNA sites occupied by a factor of interest. To do this, an antibody specifically recognizing a DNA-binding protein is used to immunoprecipitate the protein that has been cross-linked to its DNA-binding sites in living cells. Within epigenomic projects, the protein of interest mostly comprises a modified histone, although the technique can be extended to proteins that are part of the general transcription machinery, sequence-specific transcription factors, and other DNA-interacting proteins such as those involved in replication, recombination, or repair. Until recently, global identification of ChIPed DNA fragments was almost always done by hybridizing the ChIP DNA to microarrays that tile part or most of the genome being studied (ChIP-chip). Although valuable data have been generated this way, ChIP-chip suffered from limitations generally associated with microarrays such as the low spatial resolution and genomic coverage per microarray, its requirement for multiple arrays to cover a complete genome of higher eukaryotes, and the biases introduced in the ChIP-chip procedure. 15 ChIP-seq has overcome most of the biggest problems. In ChIP-seq, massive parallel sequencing of ChIP DNA generates libraries of relatively short DNA sequences that can be mapped to an annotated genome, thereby determining the localization of proteins or chromatin modifications of interest. This approach has been shown to be of significant importance as it is relatively low cost and offers the possibility to perform genome-wide analysis at single base-pair resolution. 16 ChIP-seq does not suffer from the false or uncertain signals that can result from cross-hybridization, and quantification is more accurate because counting sequence reads is “digital” rather than the continuous scale of fluorescence signal obtained with microarray hybridization. Finally, ChIP-seq can focus on a binding site at higher resolution than is typical for ChIP-chip. Indeed, a size selection step on the ChIP DNA and computational features typically allow binding sites to be assigned to regions of less than 100 bp.
An important question remains, and this concerns which histone modifications should be mapped. The definition of what constitutes an epigenome of lasting value and, consequently, which marks should be mapped is affected by many parameters such as biological/clinical relevance, suitable technology/reagents, data quality, compatibility with other efforts, and again, costs. A guideline given by the IHEC consortium (http://www.ihec-epigenomes.org/) is to map the following 6 histone modifications: H3K4me3, H3K9me3, H3K27me3, H3K27ac, H3K4me1, and H3K36me3. The presence of these marks is indicative of active promoters (H3K4me3, H3K27ac), active enhancers (H3K4me1, H3K27ac), actively transcribed genes (H3K36me3), or heterochromatin regions (H3K9me3, H3K27me3). This list can be extended with many marks, and indeed, another consortium operating under the umbrella of IHEC called BLUEPRINT (see below) aims at including several other informative marks such as H2A.z as a marker for open chromatin regions, H4K20me3 as an additional mark for heterochromatin, p300 and/or CBP as additional enhancer markers, H3K79me3 as a transcription initiation marker, H3 and H4 acetylation as markers of promoters and transcription, and RNA polymerase II as a marker of transcription activity. Undoubtedly, this list can and will be extended with the development of new reagents as well as with discovery of other relevant molecular chromatin features.
RNA-Seq
For profiling mRNA populations, microarrays have dominated for more than a decade, bringing us most of what we know about entire transcriptomes from yeast and bacteria to mouse and man. 15 However, throughout the research community, this method has recently been replaced with RNA-seq. Unlike the array-based methods, RNA-seq is quantitative and will detect exon-specific expression, hence providing important information on mRNA isoforms. In addition, RNA-seq can provide a wealth of information about gene and transcript structure as sequence reads from within exons as well as reads spanning exons will be mapped (“junction reads”). 17 Also, the seq-based methods can be allele specific through detection of sequence polymorphisms and, by high sampling, detect very low abundance RNAs unambiguously, for example, from a rare subpopulation of cells present in the sample. Finally, extending RNA-seq to paired-end reads permits systematic discovery of alternative splicing events. 18
Genome-Wide DNA Methylation Mapping
Since the advent of ultra high-throughput sequencing, several groups have begun to use this technique to help measure the methylation status of DNA at CpG clusters in the human genome. These methods use methylated DNA immunoprecipitation (MeDIP) assays coupled to sequencing (MeDIP-seq) 19 or capturing of methylated DNA followed by sequencing (MethylCap-seq). 20 These assays are aimed at producing a reduced genome sample of CpG island fragments, which in some methods are treated with bisulfite and then sequenced. Although these techniques do not allow a complete coverage of all CpGs in the human genome, they all provide a massive increase in the number of regions that can be assayed in a single, relatively cheap experiment compared to even the most efficient available assays in the pre-NGS era. The increases in sequencing capacity resulted in 2009 in the description of the first human DNA methylome at base-pair resolution, 21 marking a major step forward in uncovering the DNA methylome and setting a new standard for DNA methylome analysis. Apart from conventional DNA methylation, recently, another DNA modification was identified, DNA hydroxymethylation (5hmC),22,23 adding a new layer of epigenetic information but also adding an additional challenge for developing genome-wide technologies that can detect this mark and also discriminate between the 2 modifications.
Chromatin Accessibility Mapping
Chromosomal DNA is occluded by virtue of being wrapped into nucleosomes. This can be detected by endonuclease accessibility, and high accessibility is a distinct hallmark of the epigenome that is strongly associated with regulatory regions. Chromatin accessibility is a strong indicator of transcription factor occupancy and nucleosome remodeling that occurs at genomic regions participating in gene regulation or other nuclear processes. Therefore, over the past years, several techniques have emerged to assess the accessibility of the epigenome, including DNase I hypersensitivity, 11 FAIRE, 24 and nuclease accessibility 25 (NA) mapping. Mapping DNase I–hypersensitive (HS) sites has throughout many years been a valuable tool for identifying a wide variety of different types of regulatory elements, such as promoters, enhancers, silencers, insulators, and locus control regions. 26 The method utilizes the DNase I enzyme to selectively digest DNA depleted (presumably through the actions of transcription factors) of nucleosomes, whereas DNA regions tightly wrapped in nucleosome and higher order structures are more resistant. DNase-seq is a high-throughput method that identifies DNase I HS sites across the entire genome by capturing and sequencing DNase-digested fragments.
An alternative to the enzyme-based DNase-seq and NA-seq detection of functional in vivo regulatory elements in mammalian cells is FAIRE (formaldehyde-assisted isolation of regulatory elements). Apart from the lack of an enzymatic component, advantages of FAIRE include that it does not require additional treatments before the open chromatin state is captured and that it positively selects chromatin regions that are accessible.
Data Analysis
Apart from data generation, an important aspect includes the data storage and the primary data analysis, such as alignment of sequencing tags. Large-scale epigenomic projects generate such massive amounts of data that only a processed output can be stored. Furthermore, the data volume is only expected to increase over the coming years. Therefore, data need to be efficiently stored, and sufficient capacity needs to be in place to allow for this. Moreover, it is still common in large-scale projects for data to undergo primary analysis in disparate ways, whereby individual pipelines are utilized to generate data sets. 5 Such varieties in data handling impede integrated comparative analyses by introducing additional variability. Therefore, standard pipelines should be in place for primary analysis of the data sets that are generated wherever possible so as to allow optimal comparability of data sets and reduce the extra efforts that need to be made to align data sets from different projects. It can be anticipated that the IHEC consortium will have a leading role in coordinating these data-structuring efforts. Ultimately, alignment of NGS data sets will allow comparison of the different epigenomes, resulting in a better comprehension of the role of genetic and epigenetic variation in cell physiology. Although the field of epigenomic comparisons is not yet fully matured, conceptual and computational approaches are already being developed that address the heterogeneity and context dependence of these genome-wide epigenetic data sets.8,27
Recent, Ongoing, and Future Epigenomic Projects
The field of epigenetics has expanded over the last decade, and the initial small-scale epigenetic efforts at the beginning of the century were soon followed by the initiation of large epigenetic consortia that set out to understand the wealth of information stored in the epigenome. Most of these consortia had a strong epigenetic/ epigenomic component and focused on subsets of epigenetic modifications (such as DNA methylation) as well as on other biological aspects. With the arrival of new sequencing technologies, the aim of more recent projects has shifted toward mapping full epigenomes as the major objective. Clearly, these efforts require substantial resources, and as a result, the need for collaboration has grown. Indeed, initially, most projects were initiated at a national or continental level, while more recently, efforts have extended toward intercontinental collaborations (Fig. 3). As such, it is clear that mapping the epigenome is really a global effort.
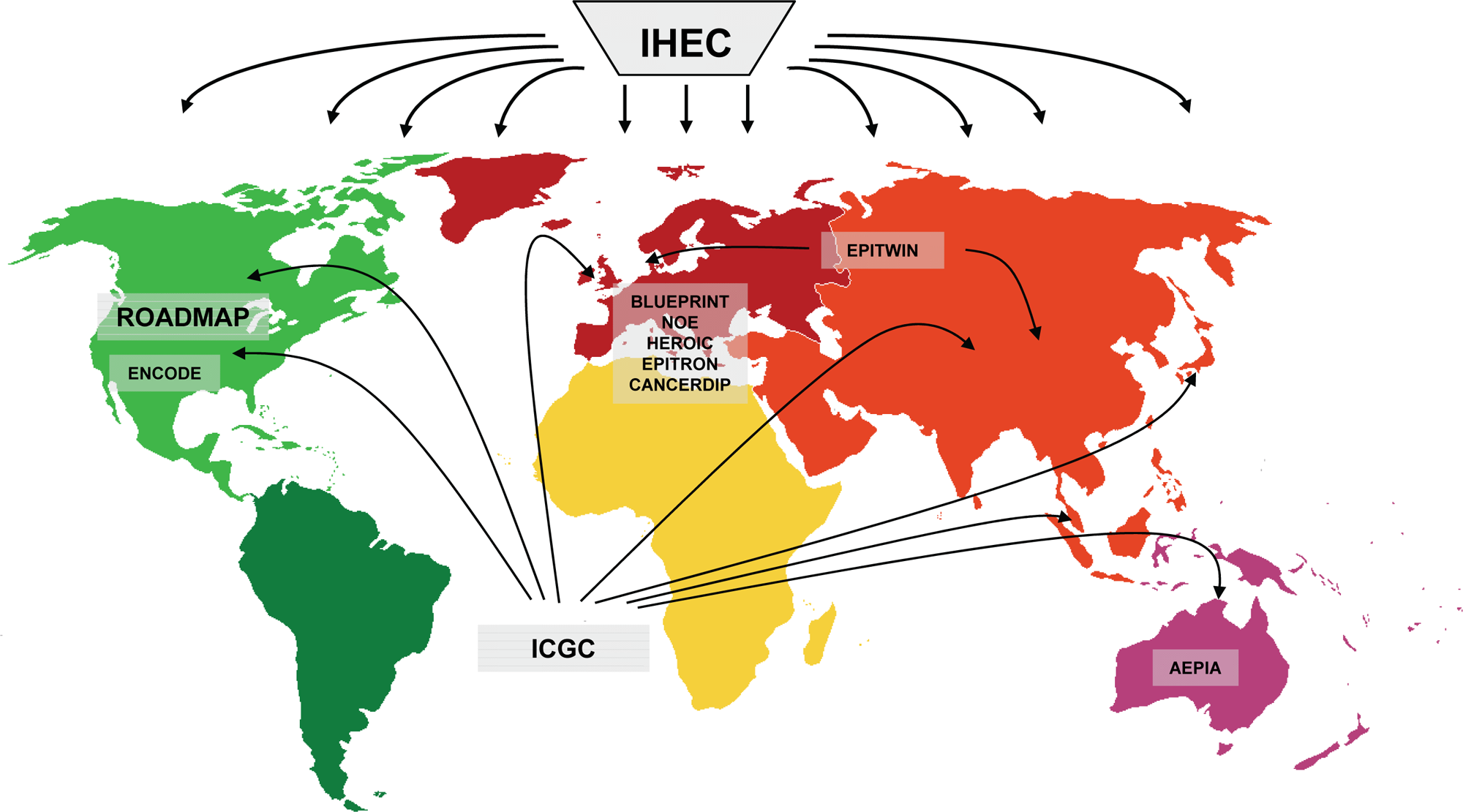
Examples of consortia that set out to map epigenomes.
One of the first projects with a major epigenetic component was the
Disease Epigenomic Projects
Many diseases are associated with alternations of the epigenome
30
and can include abnormalities caused by direct genetic changes in epigenetic enzymes as well as indirect changes through aberrant regulation of proteins regulating the epigenetic enzymes. For example, in leukemia, genetic changes in epigenetic enzymes such as the TET2
31
and EZH2
32
in myelodysplastic syndromes (MDS), MLL in acute lymphocytic leukemia (ALL) and ALL, PHF23, DNMT3A, and JARID1A in acute myeloid leukemia33,34 have been reported. In addition, in a subtype of leukemia called acute promyelocytic leukemia (APL), it was reported that a large set of epigenetic genes was targeted by PML-RARα in addition to aberrant recruitment of HDACs by this oncofusion protein,
35
showing that genetic alterations that do not directly involve an epigenetic enzyme can have a profound effect on the epigenome also. In addition, as epigenetic changes such as aberrant DNA methylation and histone modifications may be mediated by exposure to environmental or lifestyle factors, comprehensive epigenetic analysis of disease genomes is suggested to be one of the most effective ways to identify causative changes involved in tumorigenesis, irrespective of whether they are inherited or acquired. Projects such as Epitron, CancerDIP, and ICGC are examples of consortia that set out to gain more insight into the connections between aberrant regulation of epigenetic modifications and disease at a large-scale level. The
The Epigenomes of Healthy Cells
Epigenetic modifications constitute a complex layer of information on top of the genome sequence and are thought to be cell type specific. As such, every cell type and tissue that arise during development are expected to be uniquely identifiable through its epigenome. Comparisons that include varieties of pluripotent and differentiated cells or both provide powerful systems for investigating how the epigenetic code influences cellular fate. The
The
Toward a Map of All Human Healthy and Diseased Epigenomes
To coordinate all the above epigenomic efforts, a major role is set aside for the International Human Epigenome Consortium (IHEC; http://www.ihecepigenomes.org/). Its primary aim is to provide high-resolution reference epigenome maps to the research community. For this, it will coordinate epigenomic mapping and characterization worldwide to avoid redundant research efforts; to implement high data quality standards; to coordinate data storage, management, and analysis; and to provide free access to the epigenomes produced. The maps that are generated under the umbrella of IHEC should contain detailed information on DNA methylation, histone modification, nucleosome occupancy, and corresponding coding and noncoding RNA expression in different normal and disease cell types. As such, it will allow integration of different layers of epigenetic information for a wide variety of distinct cell types and thus provide a powerful resource for both basic and applied research.
What Will Be the Impact of Epigenomes?
Last February, in a series of articles published in Nature and Science to celebrate the 10-year anniversary of the human genome project (HGP), its legacy was discussed. At the time of the HGP publication, it was regarded to be the first step toward understanding and treating many human diseases. Although the HGP and follow-up sequencing efforts have yielded significant advances in many areas of basic research such as on genome anatomy, genomic variation, and human history, it is still unclear how many additional steps are needed before we actually do understand most diseases. Mapping the full epigenome of a human will be a similar accomplishment, but again, one should be careful to overestimate the short-term impact of epigenomic knowledge on understanding and curing human disease. So, on that note, what can be expected from the various epigenomic profiling projects that are currently ongoing? First and foremost, the current epigenome-wide maps will provide comprehensive lists of chromatin features that can serve as a launch point for “upstream” investigations to identify the transcription factors, regulatory molecules, and pathways that initiate, modulate, or maintain epigenomic features. Similarly, the epigenomic maps will allow pursuit of “downstream” investigations to identify genes with similar patterns of epigenetic features that suggest coordinated regulation of gene expression in particular cell types.5,8,38 Second, current epigenomic projects will establish new standards for future initiatives to produce, analyze, and integrate large data sets related to health and diseases in human and in model organisms. These include methods to optimize comparability between different data sets, avoiding duplication of effort, exploiting the economic benefits of increased mapping efforts, and developing standard operating procedures for epigenomic studies. 5 Third, because the epigenomic efforts are spread over different continents, it will be possible to compare different human populations that are subjected to different environmental settings and different nutritional behaviors. As environment and nutrition are believed to be key players in establishing the epigenome, 39 these studies will allow for the first time a comprehensive examination of the actual impact of these factors on the epigenome. Also, epigenomic mapping will have an important impact on our current understanding of many biological processes such as cellular differentiation, reprogramming, and development, as well as basic gene regulatory mechanisms. For example, mapping of DNA methylation, histone modifications, chromatin accessibility, and various species of RNA in the same cell type will provide insight into the cross-talk between different epigenetic phenomena, 40 not only at single loci, but also across larger chromosomal territories. Hence, it can be expected that these projects will reveal new principles in the regulation of genome structure and function. In terms of understanding disease and therapy design, the epigenomic maps are expected to enhance the identification of upstream factors and pathways that might contribute to a given disease state as well as downstream genes affected by the disease state.3,30 Moreover, epigenomes have great promise as discovery engines for potential biomarkers for disease states or environmental factors (e.g., toxin exposure, viral or bacterial infections, drug usage, and psychosocial stress) and thus may be useful for diagnosis or to monitor disease progression and remission. Finally, knowing what is epigenetically altered in a disease cell opens the possibility to revert the aberrant epigenetic patterns using specific drugs that would erase or override the aberrant epigenetic modification.
Still, it should be noted that, most likely, these epigenomic maps will not directly disclose all the biological functions of the genomic elements and that, in most cases, functional assays will be needed to determine the precise function and activity associated with each epigenome. Similar to the HGP project, mapping epigenomes will be a step forward toward understanding human diseases but will definitely not comprise the final step.
Outlook
It is clear that with the initiation of worldwide efforts to tackle epigenomes and the ongoing developments in sequencing, miniaturizing, and other technologies, the coming 10 years will yield information on numerous epigenomes, initially mostly focusing on human healthy and diseased tissues but gradually growing toward cells of various model organisms. These efforts will provide important new insights into the question as to the impact of cell differentiation and environmentally conditioned physiology on the epigenome. Furthermore, the availability of these data sets will provide loci where to test the causal relationship between defined epigenome features and physiology. Does the epigenome merely reflect a cell lineage’s developmental and environmental history, or does it also condition its capacity to adapt to different environments? And if so, how? Hence, the epigenomic maps will certainly advance our current understanding of normal and aberrant cellular states and will comprise another important hurdle that needed to be surmounted in the quest to acquire a full understanding of human diseases.
Footnotes
Acknowledgements
The authors thank members of the BLUEPRINT consortium for valuable input.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.