
Abstract
Tumor cells have devised several strategies to block the mitochondrial pathway of apoptosis despite endogenous or pharmacological cues to die. This process of cell death proceeds through the coordinated regulation of multiple anti-apoptotic and pro-apoptotic BCL-2 family proteins that ultimately impinge on the integrity of the outer mitochondrial membrane. Once compromised, mitochondria release pro-apoptotic factors to promote caspase activation and the apoptotic phenotype. Within the BCL-2 family exists a subclass of pro-apoptotic members termed the BH3-only proteins, which directly and/or indirectly functionally regulate the remaining anti- and pro-apoptotic BCL-2 proteins to compromise mitochondria and engage apoptosis. The focus of this review is to discuss the cellular and pharmacological regulation of the BH3-only proteins to gain a better understanding of the signaling pathways and agents that regulate this class of proteins. As the BH3-only proteins increase cellular sensitivity to pro-apoptotic agents such as chemotherapeutics, numerous small-molecule BH3 mimetics have been developed and are currently in various phases of clinical trials. Toward the end of the review, the discovery and application of the small-molecule BH3 mimetics will be discussed.
Introduction
Apoptosis is a physiological form of programmed cell death that is essential for metazoan development and tissue homeostasis. Aberrations in apoptotic signaling are implicated in numerous disease states, including autoimmunity, cancer, and degenerative disorders.1-5 Importantly, alterations within the apoptotic pathway contribute to tumorigenesis and confer resistance to not only physiological apoptotic stimuli but therapeutic regimens as well. Apoptotic signaling can initiate from outside the cell via plasma membrane receptors (referred to as the extrinsic pathway; e.g., CD95/FAS or tumor necrosis factor receptor [TNFR]) or through stress that originates from within the cell (referred to as the intrinsic or mitochondrial pathway; e.g., macromolecular damage) (Figure 1).6,7 The extrinsic apoptotic pathway is engaged when pro-apoptotic ligands such as TNFα or CD95L/FASL trigger death receptor signaling, which directly leads to the activation of caspases, which are the cysteine-aspartic proteases responsible for inducing the apoptotic phenotype (e.g., DNA laddering, loss of plasma membrane asymmetry, and cellular blebbing). 8 The intrinsic pathway is triggered by intracellular stress signals such as DNA damage, oncogenes, hypoxia, and growth factor withdrawal. These cellular stressors transcriptionally and posttranscriptionally regulate the B cell CLL/lymphoma-2 (BCL-2) family of proteins, which are responsible for mitochondrial integrity, and subsequent caspase activation and apoptosis. 9 Although these two general pathways are distinct, there are situations of crosstalk where the extrinsic pathway promotes apoptosis through the intrinsic pathway and vice versa. In these situations, the BCL-2 family of proteins is also a crucial control point of cellular fate.
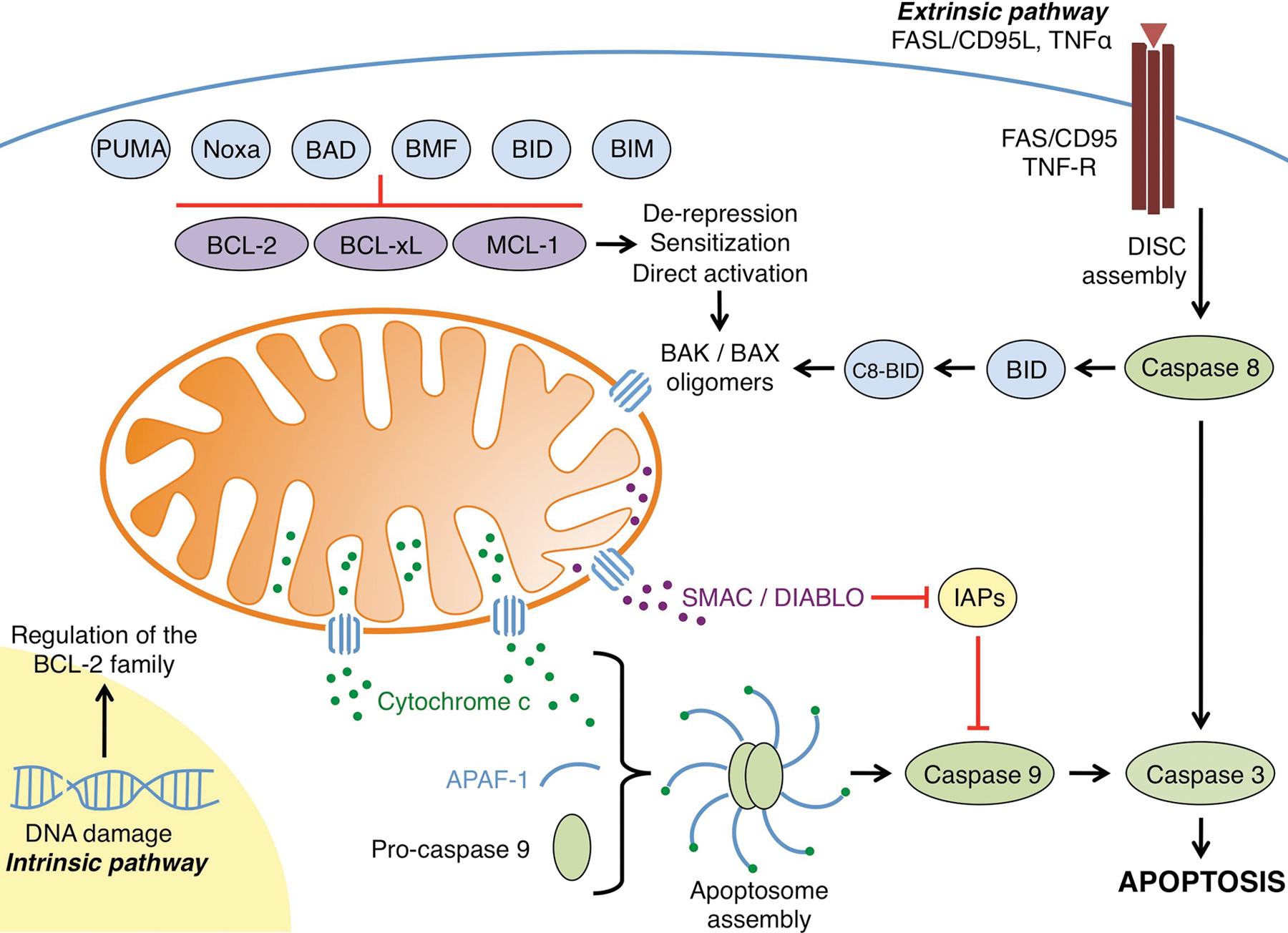
The regulation of apoptosis by the BCL-2 family. The intrinsic pathway is initiated by cellular stress signals such as DNA damage and growth factor/cytokine withdrawal. These intrinsic signals lead to the transcriptional upregulation and/or activation of pro-apoptotic BH3-only proteins such as BIM, BID, BAD, BMF, Noxa, and PUMA. These proteins are able to bind anti-apoptotic members of the family (e.g., BCL-2, BCL-xL, and/or MCL-1) and inhibit their activity. In addition to binding and inhibiting anti-apoptotic proteins, direct activator BH3-only proteins (e.g., BID and BIM) also bind and activate the effector molecules BAK and BAX. Once activated, BAK and BAX homo-oligomerize and subsequently form pores in the outer mitochondrial membrane, leading to mitochondrial outer membrane permeabilization. Pro-apoptotic proteins such as cytochrome c and SMAC/DIABLO are subsequently released from the intermembrane space into the cytosol. Cytochrome c forms a complex with APAF-1 and pro-caspase 9, whereas SMAC/DIABLO binds the inhibitors of apoptosis proteins (IAPs), which normally bind and inhibit initiator as well as effector caspases. Both steps allow for the dimerization and activation of caspase 9, subsequent cleavage and activation of effector caspase 3, and the apoptotic phenotype. The extrinsic apoptotic pathway is activated by extracellular signals such as death ligands, including FASL/CD95L and tumor necrosis factor (TNF) α. Binding of these ligands to their receptors (FAS and TNF receptor, respectively) causes receptor trimerization and subsequent recruitment of several factors that form the death-inducing signaling complex (DISC). The DISC activates initiator caspase 8, which subsequently activates effector caspase 3. Crosstalk can occur between the extrinsic and intrinsic pathways following the DISC assembly. Activated caspase 8 is able to cleave and activate the pro-apoptotic BH3-only protein BID, leading to C8-BID, which can simultaneously initiate the intrinsic apoptotic pathway through effector molecule activation.
The BCL-2 family comprises a network of related proteins that are defined by their α-helical composition of up to 4 BCL-2 homology domains (BH) and are divided into 3 classes: the anti-apoptotic BCL-2 proteins (e.g., BCL-2, BCL-xL, and MCL-1), the pro-apoptotic BCL-2 effectors (e.g., BAK and BAX), and the pro-apoptotic BH3-only proteins (e.g., BID, BIM, and PUMA) (Figure 2). 10 The founding member of the family is the anti-apoptotic protein BCL-2, which was identified as a molecular hallmark of follicular B cell lymphoma at the chromosomal translocation t (14; 18). 11 Since its discovery as an oncogene, the function of BCL-2 has been defined as preventing cell death as opposed to inducing cell proliferation. 12 Subsequently, numerous anti- and pro-apoptotic members have been identified, and each functionally cooperates within the BCL-2 family. In general, the transcriptional and posttranslational regulation of individual members, along with the numerous BCL-2 family protein-protein interactions, regulates the integrity of the outer mitochondrial membrane (OMM). Following pro-apoptotic treatment or accumulated stress, anti-apoptotic BCL-2 proteins become functionally inhibited, whereas pro-apoptotic BCL-2 proteins are enabled to permeabilize the OMM, through the process of mitochondrial outer membrane permeabilization (MOMP). MOMP occurs when proteolipid pores are created in the OMM, allowing for the release of mitochondrial proteins, such as cytochrome c and SMAC/DIABLO (second mitochondria-derived activator of caspase/direct inhibitors of apoptosis protein [IAP]–binding protein with low pI) from the mitochondrial intermembrane space into the cytosol, where they directly promote caspase activation and rapid apoptosis (Figure 1).
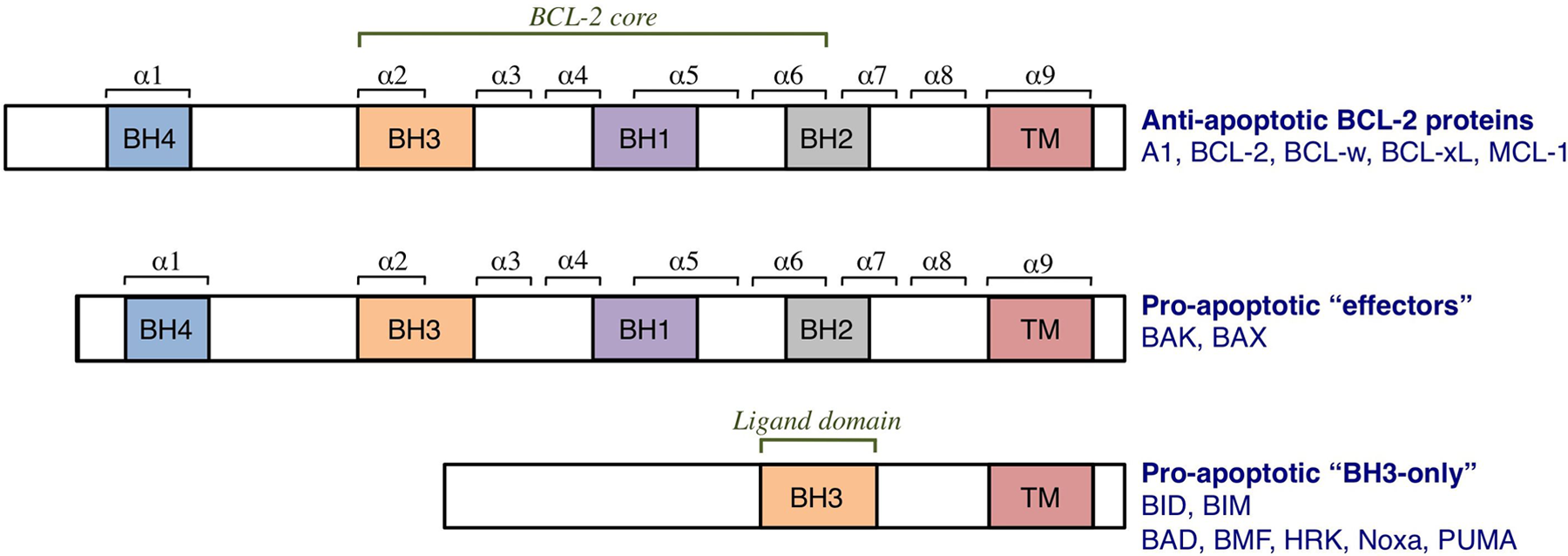
The BCL-2 family of proteins. The family is divided into anti- and pro-apoptotic members. The anti-apoptotic members, which include A1, BCL-2, BCL-xL, BCL-w, and MCL-1, share homology in all 4 BCL-2 homology (BH) domains (BH1-4). BCL-2 homology domains 1 to 3 comprise the BCL-2 structural core and create the hydrophobic groove. It is this “BCL-2 groove” that is the binding site for the BH3 domains of pro-apoptotic members. The pro-apoptotic members are subdivided into “effector” proteins and “BH3-only” proteins. The effector proteins also contain homology domains 1 to 4, whereas the BH3-only proteins contain only one BH domain, the BH3, which binds the anti-apoptotic proteins. Many BCL-2 proteins also contain a hydrophobic transmembrane domain (TM).
Here we review the basic mechanisms that control BCL-2 family function, MOMP, and apoptosis in the context of tumor cell development and resistance to anti-tumor treatments. Finally, we discuss the role of pharmacologically regulating the BCL-2 family of proteins to enhance anti-tumor strategies and patient survival.
The BCL-2 Family
The anti-apoptotic BCL-2 proteins
The anti-apoptotic BCL-2 proteins are responsible for maintaining the integrity of the OMM. They are globular proteins that contain BH domains 1 to 4 (Figure 2). The members of this subfamily include A1 (BCL-2-related gene A1), BCL-2, BCL-xL (BCL-2-related gene, long isoform), BCL-w, and MCL-1 (myeloid cell leukemia 1). These proteins are generally found at the OMM but can also be localized to the endoplasmic reticulum (ER) membrane and in the cytosol. 13 The anti-apoptotic BCL-2 proteins preserve OMM integrity by directly binding to both classes of pro-apoptotic BCL-2 proteins (i.e., the BH3-only and effector proteins), which prevents them from cooperating to induce MOMP and subsequent apoptosis. The anti-apoptotic members share a conserved “BCL-2 core” structure composed of 6 alpha (α) helices that results in the formation of a hydrophobic groove. Although the anti-apoptotic BCL-2 proteins demonstrate conserved sequence and/or structural homologies, slight variations within this groove afford unique binding profiles between the anti-apoptotic and pro-apoptotic proteins. Likewise, many of the pharmacologic inhibitors developed to target the anti-apoptotic BCL-2 proteins are able to distinguish between the anti-apoptotic members due to variations within this groove and therefore have different binding specificities to these proteins (Table 1).
BCL-2 Family Inhibitors
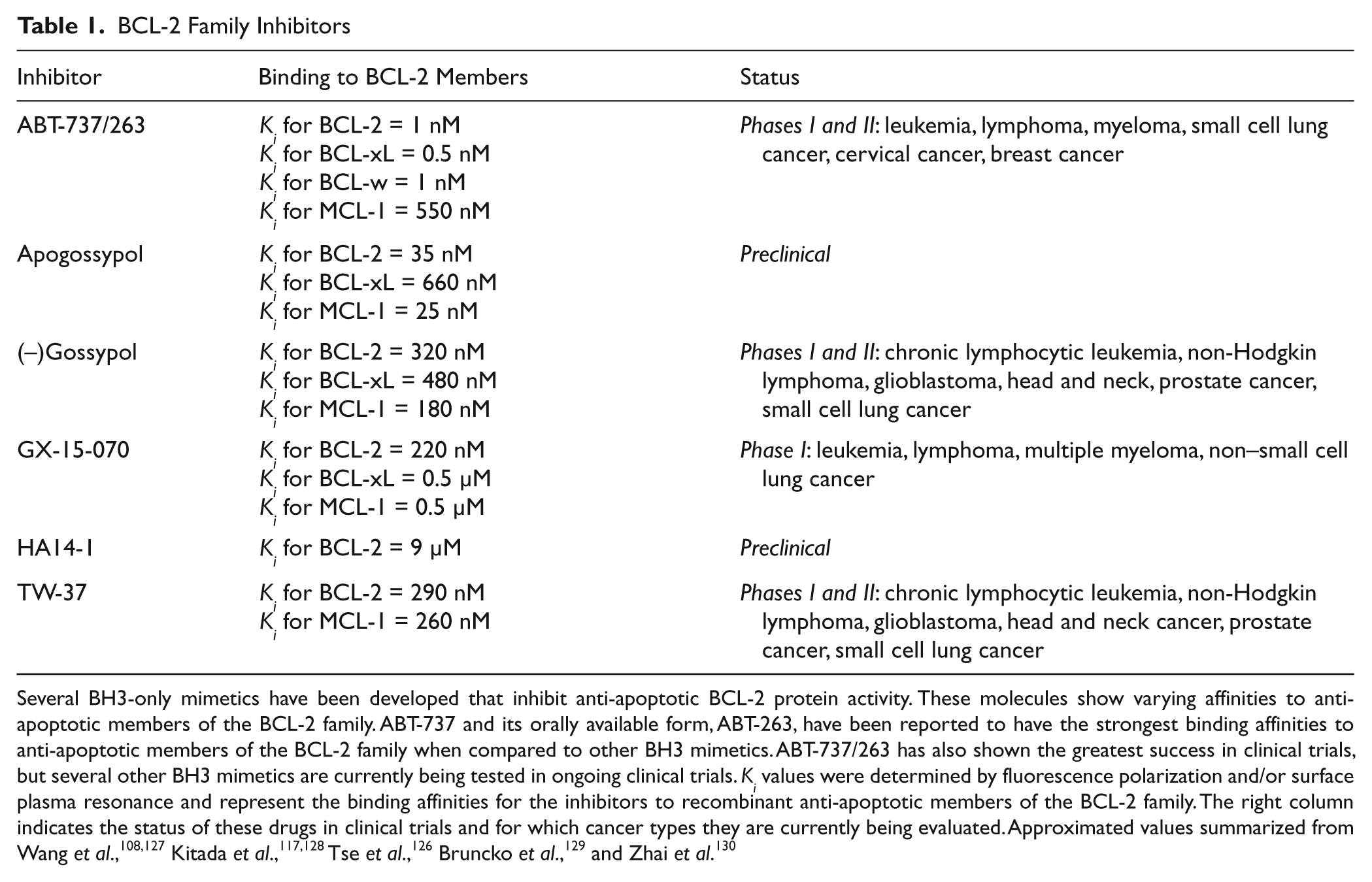
Several BH3-only mimetics have been developed that inhibit anti-apoptotic BCL-2 protein activity. These molecules show varying affinities to anti-apoptotic members of the BCL-2 family. ABT-737 and its orally available form, ABT-263, have been reported to have the strongest binding affinities to anti-apoptotic members of the BCL-2 family when compared to other BH3 mimetics. ABT-737/263 has also shown the greatest success in clinical trials, but several other BH3 mimetics are currently being tested in ongoing clinical trials. Ki values were determined by fluorescence polarization and/or surface plasma resonance and represent the binding affinities for the inhibitors to recombinant anti-apoptotic members of the BCL-2 family. The right column indicates the status of these drugs in clinical trials and for which cancer types they are currently being evaluated. Approximated values summarized from Wang et al.,108,127 Kitada et al.,117,128 Tse et al., 126 Bruncko et al., 129 and Zhai et al. 130
The pro-apoptotic BCL-2 effector proteins
The effectors are globular proteins that were originally characterized to contain BH domains 1 to 3 but recently have been suggested to also contain a BH4 domain. 14 There are two well-characterized effector proteins: BCL-2 antagonist killer 1 (BAK) and BCL-2-associated X protein (BAX), both of which can independently permeabilize the OMM to promote MOMP.15-18 Although BAK and BAX are largely redundant in their ability to affect MOMP, their localizations are different prior to pro-apoptotic stimulation. BAK is primarily localized to the OMM via a trans-membrane region in α9, but a fraction may also be found at the ER. In contrast, BAX is normally cytosolic and must re-localize to the OMM to induce MOMP. During unstressed cellular conditions, BAK and BAX exist as monomeric species, yet following sufficient pro-apoptotic stimulation, BAK and BAX “activate,” leading to their homo-oligomerization at the OMM to enable MOMP. BAK and BAX are essential for MOMP to proceed, as genetic evidence from bak–/–bax–/– animals demonstrated a complete failure to promote MOMP and marked resistance to apoptotic stimuli.19,20 Despite the opposing functions between the anti-apoptotic BCL-2 proteins and the pro-apoptotic BCL-2 effectors, they share significant sequence, domain, and structural homology. 21 As we will discuss, the activation of the effector proteins is tightly regulated by the interactions between the anti-apoptotic BCL-2 proteins and the pro-apoptotic BH3-only proteins.
The pro-apoptotic BH3-only proteins
Another subclass within the pro-apoptotic BCL-2 members is the BH3-only proteins, which include BAD (BCL-2 antagonist of cell death), BID (BH3 interacting domain death agonist), BIM (BCL-2 interacting mediator of cell death), BMF (BCL-2 modifying factor), PUMA (p53 upregulated modulator of apoptosis), and Noxa. Within this subclass, all members share homology with only a single domain, the BH3, which is defined as L-x-x-x-x-D (L = leucine, x = any amino acid, D = aspartic acid). Nearly all the BH3-only proteins are intrinsically unstructured proteins and become structured only after binding to a globular member of the BCL-2 family. 22 The major exception to the BH3-only proteins is BID, which demonstrates the conserved BCL-2 core structural motif. Despite sharing a BH3 domain, which itself is not well conserved within this subclass, the remaining flanking sequences exhibit little similarity, which may underscore the unique expression, regulation, and function of the BH3-only proteins.
The BH3-only proteins are often subdivided into two groups, depending on their ability to interact with the anti-apoptotic and effector proteins. One subgroup of BH3-only proteins, termed the direct activators, are able to bind and inhibit the anti-apoptotics but also function to bind and activate the effector proteins BAK and BAX, allowing for their oligomerization and MOMP (described below).18,23-25 The direct activator proteins include BID and BIM, and they directly engage BAK and BAX activation to induce permeabilization of mitochondrial and biochemically defined membranes. 18 Other BH3-only proteins, such as PUMA, may also demonstrate direct activation potential, but the literature is still conflicting.26,27 It is important to note that although BID and BIM are the major direct activators, the combined genetic deletion of BID and BIM results in only a minor apoptotic phenotype, suggesting the presence of either additional direct activators or alternate means of BAK/BAX activation. 28 Indeed, several reports have suggested a direct activator function for cytosolic p53, MAP-1, and ASC, and various physiochemical conditions such as heat shock can lead to BAX activation (for a comprehensive review, see Chipuk et al 9 ). It is likely that additional proteins and pathways exist that enable BAK and BAX activation and MOMP. Also of note, recent work by Ren et al. 27 demonstrated that the BH3-only proteins BID, BIM, and PUMA are essential for BAK/BAX activation, and the triple-knockout mice show a similar phenotype to that of BAK/BAX-deficient mice.
The second group of BH3-only proteins is the sensitizers/ de-repressors. Although these proteins lack the ability to directly activate BAK or BAX, they inhibit the anti-apoptotic BCL-2 network to promote MOMP. Sensitizer/de-repressor BH3-only proteins include BAD, BMF, Hrk (Harakiri), Noxa, and PUMA.29-33 These proteins bind and inhibit the hydrophobic groove of anti-apoptotic BCL-2 members, thereby preventing any future associations between the anti-apoptotics and pro-apoptotics (e.g., direct activators or effectors) (Figure 3). For example, when the hydrophobic groove of BCL-2 is unoccupied and BMF is subsequently expressed and bound to BCL-2, BMF has now “sensitized” the cell to future direct activators (and therefore BAK/BAX activation, MOMP, and apoptosis) as the BCL-2·BMF complex cannot neutralize BID or BIM activity. Another situation is when a cell harbors a complex between BCL-2 and a direct activator (e.g., BCL-2·BID); when additional BH3-only proteins are encountered, such as PUMA, they can displace BID to promote MOMP; this is referred to as “de-repression” (Figure 3). The BH3-only proteins demonstrate preferences as to which anti-apoptotic BCL-2 proteins they bind. Although BID, BIM, and PUMA are able to bind all the anti-apoptotic BCL-2 members, BAD, Noxa, and BMF are more selective within the anti-apoptotic BCL-2 repertoire. It is unlikely that one pro-apoptotic BCL-2 protein is sufficient to engage MOMP, but rather the combined interactions between the anti-apoptotic BCL-2 repertoire and the BH3-only proteins establish the threshold and trigger for BAK/BAX activation and MOMP.
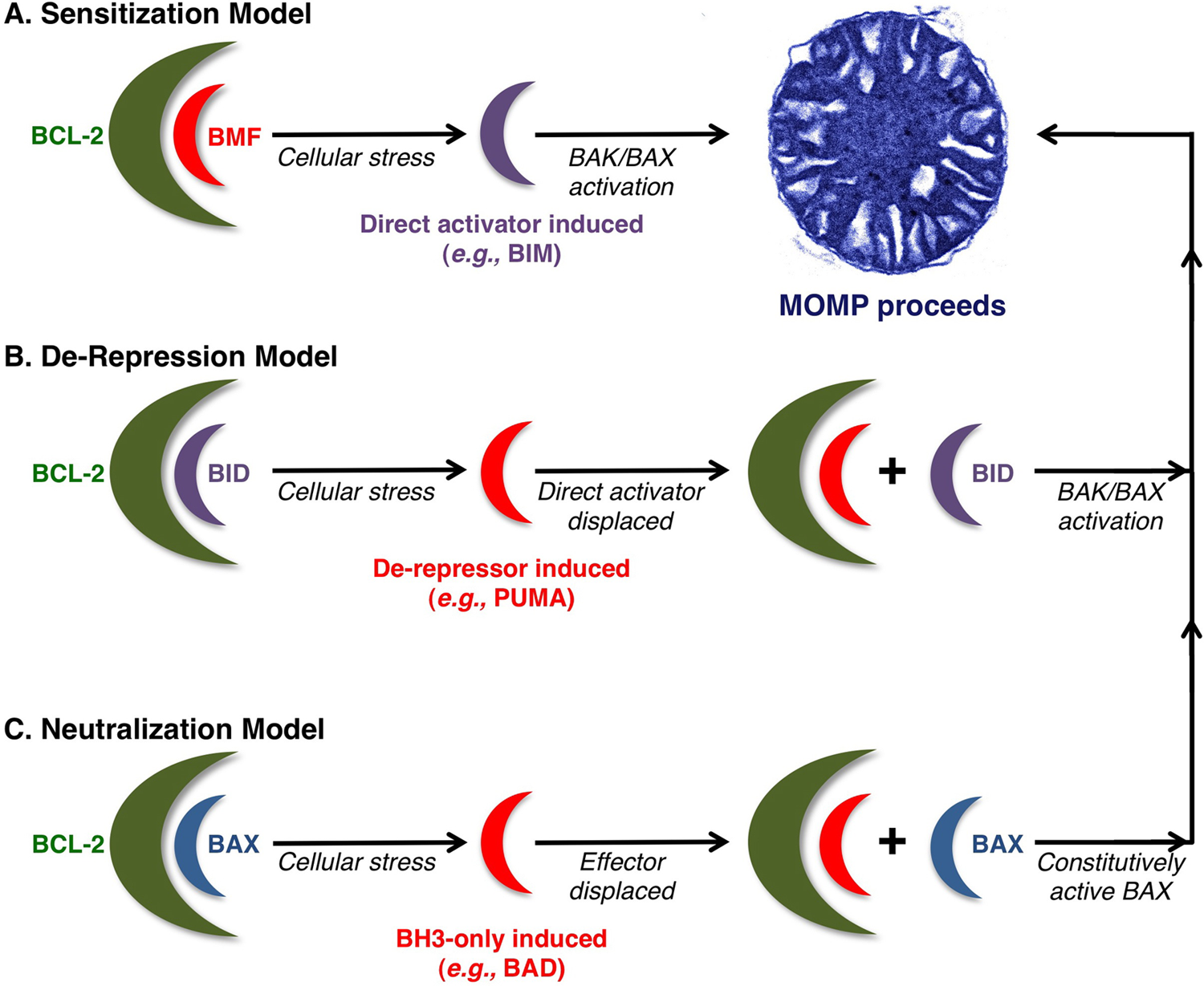
Current models for BH3-only protein functions. (
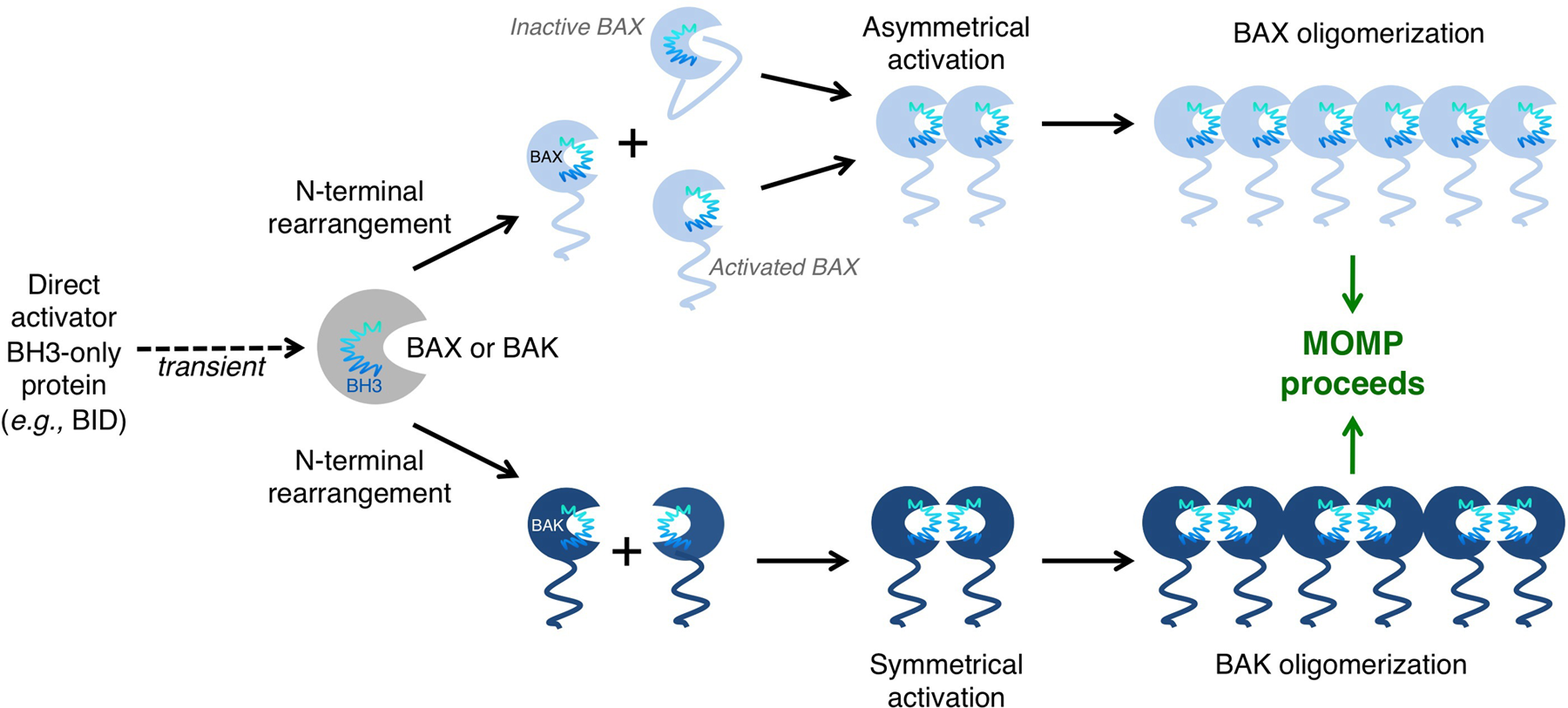
BAK/BAX activation and oligomerization promote mitochondrial outer membrane permeabilization (MOMP). BAK and BAX can be activated through interaction with direct activator BH3-only proteins and the outer mitochondrial membrane. These interactions induce conformational changes that lead to N-terminal rearrangement and the exposure of the BAK/BAX BH3 domain. In the case of BAK, rearrangement leads to the transient exposure of its BH3 domain, which subsequently binds into the hydrophobic groove of another BAK monomer. BAK dimers are able to form multimers through an α6-α6 interaction. High molecular weight species of BAK form pores in the outer mitochondrial membrane (OMM) and induce MOMP. A similar mechanism exists for BAX activation. N-terminal rearrangement of BAX leads to exposure of its BH3 domain, as well as releasing α5, α6, and α9 from the hydrophobic groove. Exposure of these α-helices allows BAX to insert into the OMM. A single BAX molecule can “propagate” this activation signal through binding of its exposed BH3 domain to inactive BAX molecules. Many activated BAX monomers form high molecular weight species once in the OMM to promote MOMP.
It is worth noting that the exact mechanism(s) by which the BH3-only proteins interact within the BCL-2 family network to promote BAK/BAX activation, MOMP, and apoptosis are still somewhat debated. There are several models explaining how the anti-apoptotic and pro-apoptotic BCL-2 family proteins interact to lead to the activation of the effectors, and these are illustrated in Figure 3. Despite these controversies, one consistent and unquestionable aspect is the indispensable role for the BH3-only proteins in regulating MOMP and apoptosis.
Activation of BAK and BAX
When coordinated direct activator and de-repressor/sensitizer BH3-only protein function has superseded the anti-apoptotic BCL-2 repertoire, the cellular response is to engage BAK and/or BAX activation, MOMP, and apoptosis. The activation of BAK and BAX is ultimately regulated through complex protein-protein interactions between the direct activator BH3-only proteins and BAK/BAX at the OMM, leading to its disruption. Recently, a better understanding of the mechanisms leading to BAK/BAX activation has emerged. As previously mentioned, BAK constitutively localizes to mitochondria, via α9 insertion within the OMM. Once activated by BID or BIM, a conformational change leads to the rearrangement of the amino-terminus, as well as subsequent exposure of the BAK BH3 domain. This exposed BH3 domain subsequently binds the hydrophobic groove of another BAK monomer, thereby forming a BAK “groove-to-groove” dimer. This dimer is able to form multimers with other BAK dimers through an α6-α6 interaction (Figure 4). 34
As discussed, BAX shuttles between the cytoplasm and mitochondria in healthy cells, which maintain mitochondrial network dynamics.35,36 Recent literature suggests that BCL-xL plays a role in BAX retro-translocation from the OMM to the cytoplasm, thereby preventing mitochondrial localization and activation of BAX. 37 However, in the presence of an apoptotic stimulus, the binding of direct activators to BAX induces amino terminal rearrangements and subsequent BH3 exposure, promoting the release of α9 from the hydrophobic groove, mitochondrial localization, and permeabilization of the OMM. It is thought that BAX α5, α6, and α9 insert into the OMM and that the activation of a single BAX molecule can propagate the activation of other BAX molecules via the exposed BAX BH3 domains to drive BAX oligomerization (Figure 4). 38 A recent publication by Kim et al. 39 supports that mitochondrial targeting and homo-oligomerization of BAX are two distinct steps in the activation of the protein, and this can be modeled in vitro. 40
The BH3-Only Proteins: A Closer Look
Cellular stress leads to the transcriptional and posttranscriptional activation of distinct BH3-only proteins, which in turn inhibits the anti-apoptotic repertoire directly, leading to BAK and BAX activation. Although the BH3-only proteins are indispensable for the mitochondrial pathway of apoptosis, evidence suggests they also regulate other cellular pathways such as metabolism, inflammation, and autophagy (for a comprehensive review, see Chipuk et al. 9 ). Integrating the multiple functions and pathways that the BH3-only proteins regulate in normal and tumor cells will provide insights into the fundamental mechanisms of this pro-apoptotic subclass of proteins and may possibly highlight their potential success to be pharmacologic targets. In the recent years, several BH3-only proteins have been studied with particular attention focused on their mechanisms of action and potential role in mediating specific signaling pathways leading to apoptosis. There is abundant evidence to suggest that the BH3-only proteins play a significant role in tumorigenesis, and this is most demonstrated in the phenotypes of knockout mice; although most of these knockout mice are developmentally normal and viable, they often do develop a malignant phenotype when challenged with a tumor-promoting background. Here we discuss the role of several BH3-only proteins in the regulation of tumor cell signaling, tumorigenesis, and treatment with an emphasis on their roles in apoptosis.
The BH3-Only Proteins and Tumor-Promoting Signaling Pathways
BAD (BCL-2 antagonist of cell death)
This BH3-only protein was originally identified by Yang et al. 33 in a yeast 2-hybrid (Y2H) screen for BCL-2 and BCL-xL binding partners. BAD is intimately regulated by cytokine and growth factor signaling and likely influences numerous aspects of metabolism, autophagy, and cell death. Since growth factor signaling is a pro-survival cue, it is important for the cell to have signaling mechanisms to promote survival and proliferation while also preventing death. 41 BAD activity is tightly regulated by kinases that negatively regulate the protein’s function through a series of phosphorylation events. Growth factor signaling leading to activation of kinases such as JNK, PKA, and AKT induces BAD phosphorylation at multiple sites (e.g., Ser112, 155, and 136).42-44 These phosphorylation events allow BAD to be sequestered by several isoforms of the 14-3-3 scaffold protein in the cytoplasm, thereby preventing its association with the anti-apoptotic BCL-2 proteins in the cytosol and OMM. It has also been suggested that phosphorylation within the BH3 domain, although not affecting its localization, does affect the interaction between BAD and the hydrophobic groove of the anti-apoptotics, rendering this interaction energetically unfavorable. 45 Mutational analysis has revealed one such residue to be S155, which lies within the BAD BH3 domain. 46 BAD is described to play a significant role in a variety of metabolic processes, including glucose metabolism and insulin secretion.47,48 It is negatively regulated by insulin growth factor signaling, suggesting that it indeed plays an active role in glucose metabolism. The phosphorylation of BAD, although negatively regulating its pro-apoptotic activity, allows it to directly interact with and activate the glucokinase protein complex, subsequently leading to changes in glucose metabolism and insulin secretion. This is an especially important point to consider in the context of tumorigenesis as the regulation of a single protein can prevent apoptosis while at the same time altering metabolism and glucose tolerance.
BAD-deficient mice are developmentally normal and viable, but the mice spontaneously develop diffuse large B cell lymphoma within 15 months of life. 49 The observation that the bad-/- mice are relatively unaffected, yet there is a propensity for tumor development in these mice, suggests that additional events take place and contribute to the tumor phenotype. This is supported by experiments where irradiation of these mice results in an accelerated tumorigenesis. 49 Furthermore, when examining the role of BAD in tumorigenesis, it is important to consider some of the pathways involved in BAD regulation since these pathways are often altered in human cancers. For example, the PI3K/AKT pathway is involved in negatively regulating BAD through an inhibitory phosphorylation at residue S136. 50 Loss of function mutations in PTEN (an inhibitor of the pathway) and gain of function in receptor signaling, leading to a constitutively active PI3K/AKT as well as Ras/MAPK pathways, have been reported in several tumor types such as breast, prostate, and glioblastoma. She et al. 51 demonstrated that in a breast epithelial model where PTEN is mutated and therefore inactive, BAD is constitutively phosphorylated and sequestered in the cytoplasm, inhibiting its pro-apoptotic activity. Since phosphorylation is the main regulatory mechanism by which cancer cells inhibit BAD function, and now several elegant studies have shed light on which pathways are involved, these data suggest that using PI3K/AKT or Ras/MAPK inhibitors in conjunction with anti-apoptotic inhibitors (discussed later) could have synergistic effects in tumors demonstrating decreased BAD expression.
BID (BH3 interacting domain death agonist)
BID is the only BH3-only protein that is mainly activated through the extrinsic death receptor pathway. This occurs via caspase-8-mediated cleavage generating the active molecule termed C8-BID. C8-BID interacts with the OMM, leading to release of the inhibitory amino terminal fragment, which may either promote the BH3-containing carboxyl-terminal fragment to insert into the OMM directly or bind to anti-apoptotic BCL-2 proteins at the OMM.40,52-54 Once BID is at the OMM, it can activate BAK and/or BAX, which drives BAK and/or BAX homo-oligomerization and the formation of MOMP-promoting pores in the membrane. 55 In addition, it has also been reported that tBID (truncated BID, which is equivalent to the carboxyl-terminal fragment) homo-oligomerizes in the OMM to promote apoptosis.56,57 It is likely that BID cleavage and activation may also be regulated by posttranslational events, such as phosphorylation by casein kinases. 58 ER stress may also induce BID cleavage through caspase-2, a potentially critical step in activating the mitochondrial apoptotic pathway in response to such stresses.59,60 Furthermore, reports have also suggested that BID can be activated through phosphorylation by ATM kinase in response to DNA damage. 61 Interestingly, it is shown that p53-mediated apoptosis in response to DNA damage can proceed through a PIDD and caspase-2-dependent mechanism, proposing an additional connection between p53- and BID-regulated tumorigenesis.62,63 However, there are conflicting results regarding the role of BID in DNA damage–induced apoptosis in vivo.61,64 Although bid–/– mice undergo normal development, they do exhibit myeloproliferative as well as leukemic disorders with age. 65 bid–/– mice, however, show no difference in apoptotic responses to DNA damage, replicative stress, or propensity for tumor development when challenged with oncogenic myc. 64 Although the role of BID in tumorigenesis remains controversial and minimal evidence supporting an influence of BID in human malignancies exists, mouse data suggest an active role for BID in tumor suppression through apoptosis or DNA damage pathways.65,66 Also of note, a recent publication by Yeretssian et al. 67 identified a role for BID in the innate immune response where BID is able to bind pattern recognition receptors, facilitating their signaling through the NF-κB and ERK pathways. bid-/- mice showed a suppressed immune response following an immunological challenge, suggesting that BID plays an active role in mediating the crosstalk between the innate immune response and apoptosis.
BIM (BCL-2 interacting mediator of cell death)
Identified by O’Conner et al. 68 as a BCL-2 binding partner, BIM is another well-characterized direct activator BH3-only protein. The protein is expressed as at least 3 different isoforms, generated through alternative splicing: BIM-EL (extra long), BIM-L (long), and BIM-S (short). The smallest of the three, BIM-S, is reported to be the most lethal of the isoforms. The protein localizes mainly to microtubules, binding to dynein light chain 1 (DLC1), and re-localizes to mitochondria in response to loss of cellular adhesion, as well as other apoptotic stimuli, presumably to bind anti-apoptotics. The pro-apoptotic activity of BIM is regulated by transcriptional as well as posttranscriptional mechanisms, some of which are the targets of chemotherapeutic drugs that aim to induce apoptosis through BIM upregulation and/or stabilization.
At the transcriptional level, BIM expression is regulated by the forkhead transcription factor, Foxo3a, which binds and activates the bim promoter in response to cytokine withdrawal. 69 Interestingly, Foxo3a is the target of several drugs, including AZD6244, which enhances Foxo3a expression, thereby increasing bim expression and inducing apoptosis. 70 bim transcription is also induced in response to ER stress. During an unfolded protein response (UPR), the transcription factor CHOP is activated and subsequently induces bim transcription. 71 BIM can also be regulated posttranscriptionally through a series of phosphorylation events. Phosphorylation can occur through a JNK-dependent mechanism, which promotes BIM dissociation from DLC1 and allowing it to re-localize to mitochondria and induce BAK/BAX activation and apoptosis. 72 Data support that inhibition of the BCR/ABL fusion oncoprotein by imatinib mesylate (an agent developed for the treatment of chronic myeloid leukemia) induces apoptosis via the activation and coordination between BIM, BAD, and BMF. It has also been shown that MEK inhibition in B-RAF mutant tumors results in apoptosis partially due to reduced phosphorylation and enhanced function of BIM. The MAPK pathway is also involved in BIM regulation as MEK inhibitors such as UO126 synergize when used in combination with BCL-2 inhibitor ABT-737 in colorectal tumor and melanoma cells expressing b-raf mutations. 73 Moreover, the treatment of lung cancer cells with EGFR tyrosine kinase inhibitors gefitinib or erlotinib showed an upregulation in bim expression, and the addition of the BH3 mimetic ABT-737 further enhanced the lethality of these drugs in non–small cell lung cancer.74,75
Decreased BIM expression is reported in several human malignancies, including B cell lymphoma and colon cancer, and is associated with poor patient survival.76,77 Interestingly, bim-/- mice develop normally with no significant incidence of spontaneous tumorigenesis; however, there is an accumulation in lymphoid and myeloid cells. 78 Also of note, in the Eµ-myc mouse model of B cell lymphoma, the loss of a single bim allele sensitizes the mice to tumorigenesis.79,80 The loss of bim in a mouse xenograft model was also found to enhance epithelial tumor growth in nude mice injected with wt or bim-/- BMK cells. 81
BMF (BCL-2 modifying factor)
The BH3-only member, BMF, was originally identified by Puthalakath et al. 32 in a Y2H screen, using the anti-apoptotic protein MCL-1 as bait. It was shown in subsequent Y2H screens that BMF interacts with the other anti-apoptotics, including BCL-2, BCL-xL, and BCL-w. Three isoforms of human BMF (BMF, BMFII, and BMFIII) have been identified and are produced from splice variants of the gene. These isoforms, however, lack the BH3 domain and hence have no effect on apoptosis, and therefore only BMFI is discussed in this context. BMF localization is regulated in a similar manner to BIM. The protein contains a conserved dynein light chain binding motif and is localized to dynein light chain 2 (DLC2), a component of the myosin V actin motor complex. 32 This association appears to be important in the regulation of pro-apoptotic activities of BMF. Once a cell loses adhesion and the actin cytoskeleton is compromised either due to anoikis or as a result of toxin treatment (e.g., cytochalasin D), BMF localization is altered, and the protein can freely bind members of the anti-apoptotic family. 32 This is especially important when considering BMF activity in hematopoietic tumors as well as metastatic solid tissue cancers, in which normal cell adhesion is altered or lost. In this same regard, BMF is upregulated at the mRNA and protein levels and is required for apoptosis during mammary epithelial morphogenesis and anoikis. 82 BMF can also be regulated epigenetically at its promoter via CpG islands: cells treated with histone deacetylase (HDAC) inhibitors show an upregulation in BMF levels, indicating that methylation of the promoter is a key negative regulatory mechanism. This becomes especially relevant in a malignant setting, especially since HDAC inhibitors are commonly used for chemotherapeutic purposes.
The role of BMF in tumorigenesis still remains to be fully elucidated, but there have been several reports suggesting that the protein may play a role in suppressing tumor development. Using the Eµ-myc model, Frenzel et al. 83 showed that when bmf is knocked out, the mice succumb to the disease much quicker. The authors also showed that loss of bmf in their model reduced the pressure for p53 loss, suggesting that BMF may be a novel component of the c-myc-driven tumor suppressor pathway. There have also been reports showing BMF upregulation during mammary acinar formation as well as anoikis, a form of programmed cell death induced in anchorage-independent cells. Studies by Schmelzle et al. 82 demonstrated that BMF shRNA in the breast epithelial MCF10A cell line resulted in an inhibition of anoikis as well as lumen formation. In their model, reducing BMF levels not only increased colony formation in vitro but also promoted anchorage-independent growth of these cells, suggesting a tumor-suppressive function of BMF.
PUMA (p53 upregulated modulator of apoptosis)
Shortly following its initial discovery, PUMA was identified as a transcriptional target of p53 through global gene expression profiling in a p53-inducible cell line, SAOS-2-p53.30,84,94 PUMA was also identified by a Y2H screen as a BCL-2 binding partner and was thus named BBC3 (BCL2–binding component 3). Once transcriptionally activated, the PUMA gene produces 2 transcripts: PUMAα and PUMAβ, both of which contain BH3 domains capable of interacting with members of the BCL-2 family. As with many of the BH3-only proteins, expression levels of PUMA are normally very low, but it is upregulated in response to several cellular stresses. One of the most well-understood transcriptional regulators of PUMA is p53. In the event of DNA damage, p53 is activated by the ATM and ATR pathways and subsequently acts as a transcription factor that directly engages the PUMA promoter and induces the production of PUMAα and PUMAβ proteins. 85 PUMA can also be activated independently of p53 and thus plays a role in p53-independent apoptosis as in the case of the p53 homolog p73, which is able to engage the PUMA promoter at the p53 response elements. In response to stresses such as growth factor/cytokine withdrawal and ER stress, PUMA is upregulated by transcription factors such as FoxO3a, C/EBP homologous protein CHOP, and E2F1.86-88 PUMA transcription can also be negatively regulated through the p53 pathway. In response to DNA damage, p53 activates the transcriptional repressor Slug, which in turn inhibits p53-mediated transcription of PUMA. 89 Recently, posttranscriptional mechanisms of regulation for PUMA have been identified. It has been reported that PUMA can be negatively regulated through phosphorylation at multiple sites, including Ser10, which promotes its proteasomal degradation. 90
PUMA possesses potent pro-apoptotic activity as a result of its ability to directly interact with all anti-apoptotic members of the BCL-2 family. PUMA mainly localizes to mitochondria (since it is bound to the anti-apoptotics) and functions to promote BAK and BAX activation through de-repression and sensitization of the anti-apoptotics sequestering them (Figure 3).26,91,92 Interestingly, recent reports suggest that PUMA can also directly activate BAK and BAX through transient binding that leads to their homo-oligomerization, although the results are in contrast to numerous reports.39,91-93 Despite playing such an indispensable role in p53-dependent and p53-independent apoptosis, puma-/- mice do develop normally and show no significant tumorigenic phenotype. 94 Also of note, inactivating mutations in puma have not been reported in any cancer type. It is worth mentioning, however, that studies performed in Eµ-myc mice showed that the loss of puma dramatically accelerates the onset of disease. The same group also reported that puma loss accelerated the time to tumor formation in E1A-transformed MEFs. 95 The combined loss of puma and bim has also been shown to accelerate disease and induce spontaneous tumor formation. 96 In terms of chemotherapeutic regulation, PUMA is targeted and subsequently induced by several kinase inhibitors. Upregulation of PUMA can be seen in melanoma cells treated with the MEK inhibitor U0126, as well as in colon cancer cells treated with the pan-kinase inhibitor staurosporine.97-99
Noxa (Latin for “damage”)
Noxa was originally identified as a phorbol myristate acetate responsive gene from an adult T cell leukemia library, but the function of the protein remained elusive. 100 A decade after its initial identification, Noxa was defined as a p53-inducible gene that encoded for a BH3-only protein that played a role in DNA damage and hypoxia-induced apoptosis.31,101 Even though Noxa does play a role in p53-mediated apoptosis, it is less essential than PUMA, possibly due to Noxa having a higher selectivity for the anti-apoptotic BCL-2 proteins (i.e., Noxa only antagonizes the activity of MCL-1, compared to PUMA, which inhibits all the anti-apoptotic BCL-2 proteins). Similarly to PUMA, Noxa can also be activated through p53-independent mechanisms. Under hypoxic conditions, the basic helix-loop-helix transcription factor HIF-1α is released from sequestration in the cytoplasm and re-localizes to the nucleus, where it transcriptionally activates the expression of several genes, including noxa. 101 The transcription factor and Rb target E2f1 has also been shown to upregulate noxa expression independently of p53.
noxa–/– mice show no obvious developmental or tumorigenic phenotype.102,103 Also of note, loss of noxa in the Eµ-myc mouse model did not accelerate tumor development, but loss of noxa did sensitize the mice to tumor formation when they were also haploinsufficient for puma. 104 Altered gene expression of noxa is reported in several human cancers, including small cell lung cancer, where Noxa overexpression is correlated with increased sensitivity to the BCL-2 inhibitor ABT-737, which, as will be discussed in further detail later, inhibits all anti-apoptotic members of the family, except MCL-1, the sole anti-apoptotic target of Noxa. 105
The BH3-Only Proteins and Chemotherapy
It is important to note that although alterations in BH3-only protein expression have been reported in several cancers, such as colon and lung cancer, this is not a common phenotype. In contrast, the regulation of anti-apoptotic members is more widely demonstrated in many cancer types, including those that show no aberrant changes in BH3-only expression. This may be a mechanism selected for by tumor cells to block the activity of several BH3-only proteins and BAK/BAX, along with additional metabolic and survival advantages conferred by increased anti-apoptotic BCL-2 protein expression. Based on these observations, anti-apoptotic members of the BCL-2 family are attractive targets for the treatment of numerous cancers. As will be discussed below, several strategies have been adopted to develop such therapies. One of the most successful attempts to create such therapeutics is evident in BH3 mimetic molecules that inhibit anti-apoptotic BCL-2 protein activity, thereby enhancing BH3-only function and promoting MOMP.
One of the first successful attempts to pharmacologically regulate the BCL-2 family was using Oblimersen, an 18-mer antisense oligonucleotide designed to target the first 6 codons of bcl-2 mRNA. 106 Despite several preclinical studies showing great promise for the ability of Oblimersen to reduce BCL-2 protein expression in tumorigenic cell lines and in mice, several clinical studies failed to reach their desired end points. 107 This may be due to Oblimersen targeting only BCL-2; it has no effect on other anti-apoptotics and may actually enhance expression and/or activity of the other proteins to overcome decreased BCL-2 expression. Given the importance of BH3-only proteins in tumorigenesis as well as their effect on drug responses, several small molecules have been developed to functionally mimic the BH3 domain (see Table 1 for a partial list). BH3 mimetics function by binding within the hydrophobic groove of the anti-apoptotic BCL-2 proteins, thereby inhibiting anti-apoptotic protein activity, and lowering the threshold or apoptosis to proceed.
HA14-1 is a compound identified via in silico screens for potentially inhibitory molecules that bind the hydrophobic groove of BCL-2. 108 After identification, HA14-1 was subsequently found to inhibit the binding of BCL-2 and BCL-xL to BAK and BAX, as well as the direct activator BIM. In subsequent years, it was shown to induce apoptosis in leukemic and lymphocytic cells as well as sensitize glioblastoma cells to apoptosis.109,110 Despite these findings, however, it has been demonstrated that BAK/BAX-deficient cells are just as sensitive to treatment with HA14-1 as wild-type cells, potentially suggesting BAK/BAX-independent mechanisms of action.
In 2007, Gemin X published the first pan anti-apoptotic BCL-2 protein inhibitor, Obatoclax. 111 This was one of the first compounds designed that not only inhibited BCL-2 and BCL-xL but MCL-1 and presumably other anti-apoptotics as well—possibly the reason it conferred low resistance in patients. Obatoclax entered into clinical trials and was found to be well tolerated in patients with hematological and myeloid malignancies.112,113 However, the exact mechanism of action of this compound has recently been called into question by several studies. Similar to reports regarding HA14-1, Konopleva et al. 114 and Mott et al. 115 both reported only slight decreases in cell death levels in bak–/–bax–/– cells (compared to wild-type cells) in response to treatment with Obatoclax. One group also showed a cell cycle arrest phenotype in response to Obatoclax treatment. Also of note, a clear correlation has yet to be shown linking increased expression of BCL-2 and other anti-apoptotics in tumors and their sensitivity to Obatoclax. These findings suggest that this compound may have BCL-2 independent targets. Although it is clear that Obatoclax does target the BCL-2 family, whether this is the major mechanism of anti-tumor activity has yet to be fully supported.
BH3 mimetics have also been derived from natural molecules, as is the case with gossypol. It is a natural polyphenol derived from the cottonseed plant. It was historically used as a male contraceptive, but due to reports of permanent infertility as well as high toxicity, further studies in the context of contraception were abandoned. Following this, several labs began studying the potential effects of gossypol on cancer, and it was subsequently found that the (–) enantiomer exhibits stronger apoptotic effects compared to the (+) enantiomer. 116 As a BH3 mimetic, (–) gossypol is able to bind BCL-2, BCL-xL, and MCL-1 although with affinities that are much lower than of the BH3-only proteins to these proteins. Gossypol derivatives have been generated, and these include Apogossypol, TM-106, and TW-37. These derivatives, TW-37 in particular, exhibit stronger binding affinities to the anti-apoptotics while also conferring less toxicity.117-119 TW-37 was the first BCL-2 inhibitor to enter the clinic. It has shown promise in inducing apoptosis and overcoming resistance in cases of B cell lymphoma, chronic lymphocytic leukemia, and multiple myeloma. 120
Despite these many attempts to generate potent BH3 mimetics, none have proven as effective at binding the anti-apoptotics as the BH3-only proteins themselves. Given the importance of the BH3 domain in the interaction between the pro-apoptotic proteins and the anti-apoptotics, Letai et al. 25 generated synthetic peptides mimicking the different BH3-only proteins. They found that peptides of BH3 domains from different proteins did not all perform the same functions, but the individual peptides did perform similar functions to the proteins they mimicked. The BH3 peptides representing BID and BIM were able to induce cytochrome c through BAK and BAX oligomerization, whereas the peptides mimicking BAD did so by binding BCL-2. One issue that arose with using synthetic peptides to bind the hydrophobic groove of anti-apoptotic proteins is the loss of α-helicity that occurs when a peptide is synthetically produced, without the remainder of the protein. The α-helical nature of the BH3 domain is what facilitates protein-protein interactions between the BH3-only proteins and the anti-apoptotics; it is that α-helix that binds to the hydrophobic groove. When a peptide is produced out of the context of the entire protein, it loses much of its structure and therefore its α-helicity. In 2004, Walensky et al. 121 pioneered a method whereby they use nonnatural amino acids containing olefin-bearing tethers to create a staple that attaches to the BH3 peptide, “stapling” it in its α-helical form. Over many studies, they generated a stabilized α-helix of BCL-2 domains (SAHBs) mimicking the BH3 domains of almost all members of the BCL-2 family. This technique of stapling the peptide not only allows it to maintain its structure and travel freely across membranes but also protects it from degradation. Not only has this technique introduced a novel strategy for testing protein-protein interactions, but it may serve as a new tool for designing cancer therapeutics to target the BCL-2 family and enhance endogenous BH3-only activity. A SAHB of the BH3 domain from the direct activator BID was shown by the same group to induce apoptosis in leukemia cells as well as inhibit growth of human leukemia xenografts in vivo. 121 One of their most recent developments is an MCL-1 BH3 SAHB, which they have shown specifically targets the MCL-1 protein to very high affinities and has also been shown to sensitize cancer cells to caspase-dependent apoptosis. 122 This identification of an exclusive and potent MCL-1 inhibitor is an important one, given the lack of current inhibitors that target MCL-1 and the emergence of MCL-1 as a chemoresistance factor in a broad range of human cancers. The development of peptides that mimic the BH3 domains of BCL-2 family members offers a much-needed tool for studying the exact nature of protein-protein interactions within the family and also provide models for developing new therapeutics to target individual proteins.
ABT-737, ABT-263
The most successful and potent BH3 mimetic compound generated to date is a compound developed by Abbott, termed ABT-737. In 2005, Oltersdorf et al. 123 used structure-activity relationship by nuclear magnetic resonance to screen a chemical library for BH3-like analogues with high binding efficiency to the hydrophobic groove of BCL-xL, generating ABT-737. The compound’s activity most closely resembles that of the BAD BH3; it is able to bind with high affinity to BCL-2, BCL-w, and BCL-xL (but not MCL-1). Accordingly, resistance to ABT-737 is reported in tumors that overexpress MCL-1. As reported by van Delft et al., 124 when compared to other BH3 mimetics that have been developed (and discussed previously), ABT-737 activity most closely resembled that of the BH3-only proteins. Importantly, unlike most of the previous BH3 mimetics, ABT-737-induced apoptosis is BAK/BAX dependent, as evidenced by the finding that BAK/BAX DKO MEFs are resistant to the treatment. 124 This study also highlighted the significance of MCL-1 in conferring resistance to treatment with ABT-737. When used in combination with downregulated MCL-1, ABT-737 is able to successfully sensitize cells to apoptosis. These results indicate that ABT-737 is the first BH3 mimetic whose functions truly mimic the BH3-only proteins. It is also the first mimetic whose mechanism of action is shown to clearly be BAK/BAX dependent. ABT-737 is well tolerated in vivo, but it has been shown to induce apoptosis in platelets, which heavily depend on BCL-xL for their survival. 125 Despite this, however, ABT-737 still shows minimal toxicity when compared to other BCL-2 inhibitors that have been entered into clinical trials. Its low toxicity and high specificity make it an ideal therapeutic especially in tumors with low MCL-1 levels or in conjunction with MCL-1 inhibitors to treat tumors overexpressing MCL-1. For use in the clinic, an orally available form of ABT-737 was developed: ABT-263. In tumor xenograft models, orally administered ABT-263 is shown to induce tumor regression in models of small–cell lung cancer and acute lymphoblastic leukemia. 126 Several phase I and phase II trials are currently under way evaluating the effect of ABT-263 on patients with malignancies of lymphoid origin.
Perspectives
Our discussions centered on how the BCL-2 family of proteins functions to regulate tumorigenesis, with a particular emphasis on BH3-only protein signaling. As the BCL-2 family defines the balance between life and death, a modern understanding of the cellular mechanisms that control its function is crucial to establish how stressed cells undergo apoptosis—and how cancer cells resist pro-apoptotic signals to ensure survival. Throughout, we suggest that numerous BCL-2 family members functionally cooperate to maintain cellular survival, and similar multipartner interactions are essential for apoptosis to proceed. These interactions appear to be directly regulated by numerous signaling cascades, many of which are commonly abrogated in cancer cells. From this, it comes as no surprise that inhibiting the cellular signaling pathways that promote survival along with lowering the threshold for apoptosis to proceed represents the ideal regimen for pharmacologically targeting cancer cells. The development of small-molecule inhibitors to the anti-apoptotic BCL-2 proteins is a milestone in modern chemotherapeutic strategies, and complementing these drugs with specific drugs to silence pro-survival pathways is already showing signs of clinical success. As we further dissect the mechanisms of the BCL-2 family and apoptotic pathways, novel therapeutic targets will certainly emerge and it is hoped provide further benefits to patients.
Footnotes
Acknowledgements
We apologize for not being able to discuss and cite all the relevant literature due to space restrictions. We would like to thank members of the Chipuk Laboratory for useful discussions and feedback on this review.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The laboratory is funded by the National Institutes of Health [grant R01CA157740] and is supported in part by a research grant from the March of Dimes Foundation [5-FY11-74].