
Abstract
Hematopoiesis is the cumulative result of intricately regulated signaling pathways that are mediated by cytokines and their receptors. Studies conducted over the past 10 to 15 years have revealed that hematopoietic cytokine receptor signaling is largely mediated by a family of tyrosine kinases termed Janus kinases (JAKs) and their downstream transcription factors, termed STATs (signal transducers and activators of transcription). Aberrations in these pathways, such as those caused by the recently identified JAK2V617F mutation and translocations of the JAK2 gene, are underlying causes of leukemias and other myeloproliferative disorders. This review discusses the role of JAK/STAT signaling in normal hematopoiesis as well as genetic abnormalities associated with myeloproliferative and myelodisplastic syndromes. This review also summarizes the status of several small molecule JAK2 inhibitors that are currently at various stages of clinical development. Several of these compounds appear to improve the quality of life of patients with myeloproliferative disorders by palliation of disease-related symptoms. However, to date, these agents do not seem to significantly affect bone marrow fibrosis, alter marrow histopathology, reverse cytopenias, reduce red cell transfusion requirements, or significantly reduce allele burden. These results suggest the possibility that additional mutational events might be associated with the development of these neoplasms, and indicate the need for combination therapies as the nature and significance of these additional molecular events is better understood.
A Basic Overview of Cytokine Receptors
Cytokines are defined as a group of growth factors that bind to their cognate receptors and trigger intracellular signaling events that result in the modulation of gene expression. Most cytokine receptors are comprised of a multi-subunit complex containing a unique and specific ligand binding subunit and a signal transducing subunit, with the latter sometimes being structurally similar to other members of the cytokine receptor superfamily 1-4 (Fig. 1). The average extracellular domain is comprised of approximately 210 amino acids and contains one or more conserved cysteine (C) residues. A second conserved sequence of tryptophan-serine-X-tryptophan-serine (W-S-X-W-S) is also present in the carboxy-terminus. 5 Structurally, the C-terminus is composed of 2 fibronectin type III modules that are connected by a hinge region, and also contains the W-S-X-W-S motif that is predicted to function as a ligand interaction domain. The cytoplasmic domains of cytokine receptors share a limited level of similarity in their membrane proximal regions, particularly in regions termed the box 1 or the proline-rich motif and the box 2 motif. These portions of the receptor are critical for proper function of the receptor and mediating mitogenic signals. In contrast, the membrane distal region, in which the box 1 and 2 motifs are located, is required for differentiation. Functions of regions within the cytoplasmic domains that do not contain box 1 or box 2 motifs vary from receptor to receptor (reviewed in Ihle et al. 6 and Vainchenker et al. 7 ).
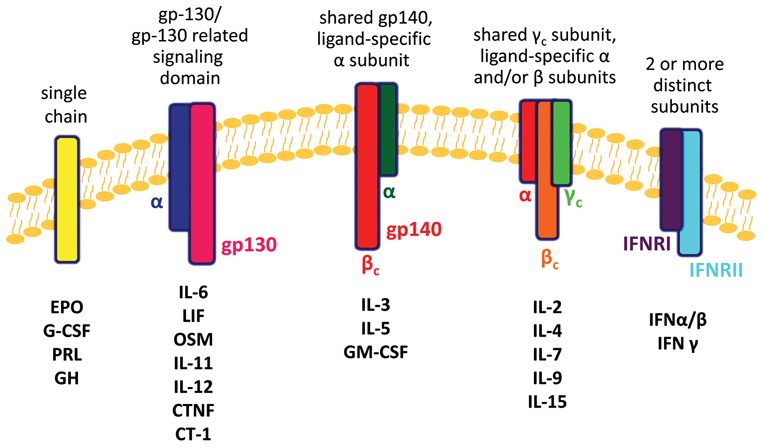
Schematic representation of cytokine receptor families. The structure of 5 superfamilies and their respective members are depicted. Note: not drawn to scale. Adapted from Baker et al. 8
There are 2 cytokine superfamilies that can be further subdivided into multiple subgroups of cytokine receptors based on the nature of shared subunits, and all are devoid of catalytic activity (reviewed in Baker et al. 8 ). Nevertheless, interaction of a cytokine with its receptor rapidly induces tyrosine phosphorylation of the receptor and a variety of cellular proteins. In the early 1990s, genetic complementation studies in an interferon-alpha (IFN-α) unresponsive cell line showed that TYK2, an “orphan” tyrosine kinase, could rescue the mutant phenotype. 9 Within a year, several additional studies were published that demonstrated that other members of the Janus kinase (JAK) family of tyrosine kinases mediated the signals triggered by cytokines. 10-13
Janus Kinases
There are 4 members of the JAK family: JAK1, JAK2, JAK3, and TYK2. Of these, TYK2 was the first to be discovered using a degenerate oligonucleotide-based screen aimed at isolating novel tyrosine kinases. 14 Shortly thereafter, cDNAs encoding JAK1, 15 JAK2, 11,16 and JAK3 17-19 were isolated using various cloning strategies.
The unique structure of the JAK kinases clearly distinguishes them from other members of the protein tyrosine kinase family (Fig. 2). The amino-terminal half of JAKs contains 2 motifs, a Src homology-2 (SH2) domain and a band four-point-one, ezrin, radixin, moesin (FERM) domain. The SH2 domain encompasses what was originally defined as the JH3 and part of the JH4 domains. However, in spite of the homology to SH2 sequences, this region does not appear to bind to phosphotyrosine residues. 20-22 Furthermore, mutation of a residue that is normally detrimental to SH2 domain function does not affect cytokine receptor activation. These studies suggest that the JAK family SH2 domains do not function as traditional SH2 domains and may, in fact, play roles as scaffolds. 23
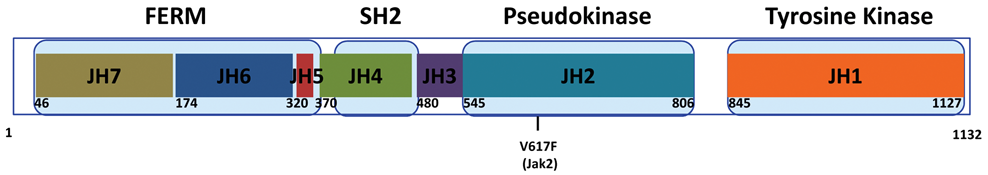
Structure of Janus kinases (JAKs). JAKs harbor 4 functional domains: the FERM domain, the SH2 domain, the pseudotyrosine kinase domain, and a catalytically active tyrosine kinase domain. Although the JH2 region is not a functional tyrosine kinase, this domain negatively regulates the activity of JAK proteins. Also shown are the FERM and SH2 domains, which serve as receptor association and phosphotyrosine binding domains, respectively, and the JAK2V617F mutation that is found in MPDs. Depicted here are the domain boundaries for JAK2. Adapted from Baker et al. 8
The regions previously termed the JH5 to JH7 (as well as part of the JH4) domains comprise the FERM domain, which regulates catalytic activity and mediates association with receptors and other proteins. An intact FERM domain is required for activated mutants of JAK1 to support type I IFN signaling. 24 In addition, mutation of tyrosine 913 in the JAK2 FERM domain has also been shown to result in constitutive activation of the kinase in the absence of cytokine stimulation. 25 Patient-derived JAK3 proteins with mutations in the FERM domain illustrate the significance of this domain in the protein’s function. These proteins were shown to be devoid of kinase activity (partly due to the fact that the kinase and FERM domains interact) and also failed to associate with receptors. 26 Conversely, chimeric proteins that contain only the JAK3 FERM domain associate with the common gamma (γc) subunit. 27 Residues located in the JH7 region have been shown to mediate the binding of JAKs to the box 1/proline-rich region of cytokine receptors, 28-30 and this interaction ultimately regulates receptor localization and turnover. 31-33
Nevertheless, the most intriguing and unique feature of these proteins is the presence of 2 Jak homology (JH) domains, JH1 and JH2, which have extensive homology to tyrosine kinase domains. The presence of the 2 kinase domains is actually the basis for the name of the protein family, being named after Janus, the Roman god with 2 faces. 15 Although the JH1 domain is a functional tyrosine kinase domain with a requisite YY motif in the activation loop, 34-36 the JH2 domain, despite harboring most of the conserved amino acids that are a characteristic of functional kinases, lacks any observable tyrosine kinase activity due to the absence of residues that are required for catalytic activity and nucleotide binding. However, it is now clear that this kinase-like domain plays a significant regulatory role in both the activity of JAK family proteins and cytokine-induced signaling. An early theoretical model of the JH1 and JH2 domains strongly suggested that the 2 domains interact with each other and that the JH2 domain exerts a negative effect on the kinase activity of the JH1 domain. 37 Subsequent biochemical studies demonstrated that both the JAK2 and JAK3 JH2 domains negatively regulate the proteins’ kinase activities. 38,39
The importance of the JH2 domain in the regulation of JAK activity has been underscored in patients suffering from myeloproliferative neoplasms (MPNs), in particular, those whose cells express the mutant JAK2V617F protein. 40-43 This mutation disrupts the inhibitory role that the JH2 domain has on JH1, whereby the activation loop of JH1 adopts a conformation such that it can be phosphorylated by an adjacent JAK2V617F molecule. The consequences of this mutation at the molecular level and its clinical significance are discussed in greater detail in subsequent sections of this article.
Cytokine Receptor Signaling via JAKs
JAKs mediate signals from a variety of cytokines and growth factors. In general, receptor dimerization/oligomerization due to ligand binding results in the juxtapositioning of the JAKs, either through homodimeric or heterodimeric interactions. The recruitment of JAKs results in their phosphorylation, either via autophosphorylation and/or transphosphorylation by other JAKs or other families of tyrosine kinases. This activation results in increased JAK kinase activity. Activated JAKs then phosphorylate receptors on target tyrosine residues that serve as docking sites that allow the binding of other SH2 domain–containing signaling molecules such as STATs, Src kinases, protein phosphatases, and other adaptor signaling proteins such as Shc, Grb2, and PI-3 kinase (reviewed in Baker et al. 8 ) (Fig. 3).
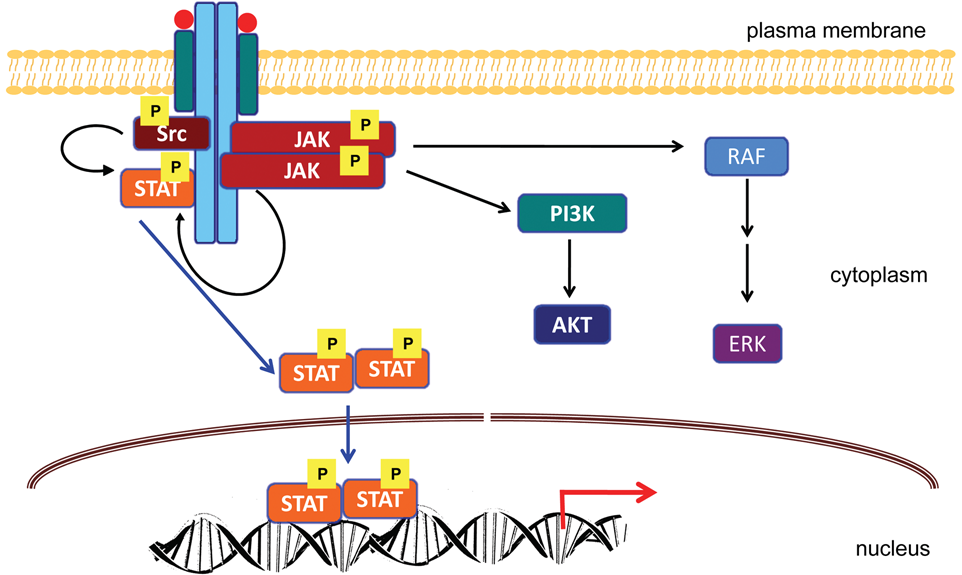
Signaling pathways mediated by JAKs. Ligand binding to cytokine receptors induces their dimerization. JAKs, which bind to the receptor via their SH2 domains, undergo transphosphorylation and subsequently phosphorylate STATs. The activated STATs dimerize and translocate to the nucleus, whereby they activate or repress target gene promoters. STATs can also be directly activated by Src kinases. In this model, JAKs phosphorylate the receptor and create binding sites for STATs. In addition to JAKs and STATs, cytokine receptors also activate additional signaling pathways involving proteins such as Akt and ERK. Adapted from Baker et al. 8
The above model is supported by several studies in which investigators have demonstrated the ability of cytokines to rapidly induce receptor oligomerization leading to JAK2 activation. 10,13,14 Furthermore, studies using chimeric receptors with various combinations of extracellular ligand binding and cytoplasmic domains also favor this model, 44-46 which readily applies to both single chain receptors such as those for EPO, PRL, GH, and G-CSF as well as multichain receptors such as those for IL-3, IL-5, and GM-CSF. Cytokine binding results in the association of the JAKs with one of the receptor subunits. The receptor-associated JAK kinase can then either process the signal or, subsequent to receptor oligomerization, can recruit other JAKs in the vicinity. Homodimerization or heterodimerization of the JAKs, followed by their activation upon phosphorylation, finally results in the propagation of the initial signal and ultimately leads to the activation of transcription factors.
Signal Transducers and Activators of Transcription
Signal transducers and activators of transcription, or STATs, are transcription factors that were originally described by Darnell et al. 47,48 as ligand-induced transcription factors in IFN-treated cells. Subsequent studies by a number of groups showed that STATs play critical roles in signal transduction pathways triggered by several cytokines and growth factors (reviewed in Murray 49 ). To date, 7 mammalian STAT-encoding genes have been identified, and alternative splicing or posttranslational proteolytic cleavage generates additional forms of STATs 1 and 3. 50 STAT-4 also exists in 2 forms, termed STAT-4α and STAT-4β, and 2 STAT-5 isoforms, termed STAT-5a and STAT-5b, are encoded by separate genes that are linked in tandem. 51,52
Like most transcription factors, STATs exhibit a modular structure with 7 well-defined domains, including an N-terminal conserved domain, a coiled-coil domain, a DNA binding domain, a linker region, an SH2 domain, a tyrosine activation, and a C-terminal transactivation domain (Fig. 4). The amino-terminal region of STATs is well conserved among family members and is critical for STAT function, as small deletions in this region have been shown to eliminate the ability of STATs to be phosphorylated. This domain also functions in nuclear import, export, receptor binding and cooperates with the DNA binding domain. The amino-terminal region also regulates dimerization of STATs in their inactive state 53 (reviewed in Schindler et al. 54 ). The coiled-coil domain adopts an α-helical conformation, functions in receptor binding, and associates with regulatory proteins (reviewed in Kisseleva et al. 55 ). The DNA binding domain is also highly conserved among the STATs, and all STAT homodimers with the exception of STAT-2 differentially bind more than 10 related γ-activated sequence (GAS) elements that are characterized by the consensus sequence, TTNCNNNAA. 56,57 A complex comprised of STAT1, STAT2, and IFN regulatory factor (IRF) 9 binds to the IFN-α/β–stimulated response element (ISRE) (AGTTN3TTTC) (reviewed in O’Shea et al. 58 ). The linker domain functions as a spacer to maintain proper conformation between the dimerization and DNA binding domains. The SH2 domain, which is the most highly conserved domain among the STATs, plays a very important role in STAT signaling, being critical for the recruitment of STATs to activated receptor complexes and for the interaction with JAK and Src kinases. In addition, this domain is required for STAT homodimerization and heterodimerization, which in turn appears to be critical for nuclear localization and DNA binding activities. The transactivation domain varies among family members and, as the name implies, modulates the transcriptional activation of target genes. C-terminally truncated isoforms of STATs 3, 4, and 5 are missing portions of their transactivation domains and reportedly behave as dominant-negative proteins (reviewed in Schindler and Strehlow 59 ).
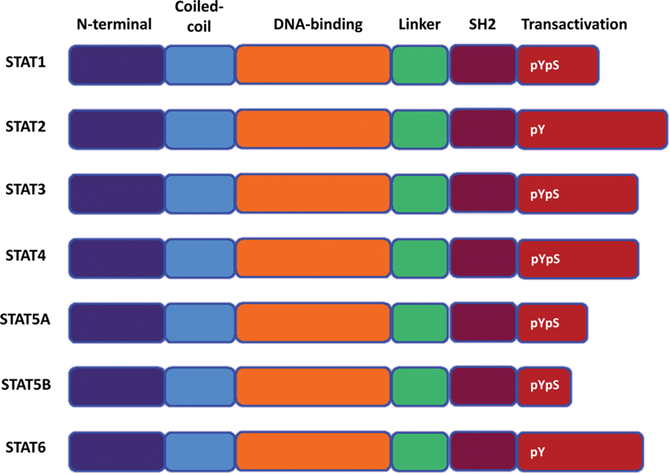
Structure of STAT proteins. STATs harbor 6 domains: the N-terminal, coiled-coil, DNA binding, linker, SH2, and transactivation domains. The N-terminal and DNA binding domains cooperate in binding to the promoters of target genes. Regulatory phosphotyrosine (pY) and phosphoserine residues (pS) are also shown. Adapted from Baker et al. 8
In an unstimulated cell, STATs are inactive, cytosolic proteins that exist in an unphosphorylated state. Cytokine stimulation induces phosphorylation of tyrosine residues on the receptor that serve as docking sites for STATs via their SH2 domains. Once bound to the receptor, all members of the STAT family become tyrosine phosphorylated in response to cytokine stimulation at a conserved carboxy-terminal tyrosine, Y694, for example, in the case of STAT-5. Phosphorylation of this tyrosine residue appears to be achieved by growth factor receptors as well as JAK and Src kinases, depending on the cell type and the nature of the ligand/receptor interactions. This form of phosphorylation induces reorientation of the STAT proteins and homodimerization and heterodimerization via the interaction of the SH2 domain of one STAT molecule with the phosphotyrosine residue of another. Once phosphorylated, the dimerized STATs translocate to the nucleus. In addition to tyrosine phosphorylation, all STATs, with the exception of STAT-2, are regulated by serine phosphorylation at a conserved PSMP motif that is located in the transactivation domain. C-terminal serine phosphorylation is stimulated by several cytokines and is mediated by serine/threonine kinases including, but not limited to, ERK, p38, JNK, mTOR, NLK, CaMKII, IKKε, and PKCδ and positively regulates the transactivation potential of these proteins (reviewed in Schindler et al. 54 and Khwaja 60 ).
Cytokine Signaling and STAT Activation by JAKs and Src Family Kinases
Because the cytokine receptor-ligand interactions result in the activation of JAK kinases that often exist in association with cytokine receptors, and because this activation is obligatory for the activation of STATs, it is widely accepted that STATs are substrates for JAK kinases. However, activated JAK kinases do not seem to exhibit specificity for a particular STAT, as different receptors activate a common STAT even though they activate distinctively different JAKs. 48,61 In addition, chimeric receptor molecules that harbor different JAK binding sites but identical STAT binding sites can activate the same STAT protein(s). 61,62 Thus, the specificity for STAT phosphorylation appears to be dictated by the docking sites for STATs that are present in the receptors themselves.
The notion that STATs are activated by kinases other than JAKs was at first demonstrated by studies aimed at investigating the molecular mechanisms associated with Src-mediated transformation. v-Src–transformed NIH3T3 cells constitutively express tyrosine-phosphorylated STAT-3, 63,64 and in vitro studies have shown that v-Src can bind to and phosphorylate STAT-3. 64 Similarly, v-Src–transformed 32Dcl3 myeloblastic cells constitutively express phosphorylated forms of STAT-1, -3, and -5 in the absence of cytokine. 65 In this model, STAT-3 activation is blocked by a dominant-negative mutant of Src, but not that of JAK-2. 66 These events mirror the signaling events induced by IL-3 stimulation, whereby the same STATs are activated and endogenous c-Src associates with and mediates the activation of STAT-3.
Based on these results, a second model of STAT activation has been proposed, where JAK kinases may be more crucial to the phosphorylation of cytokine/growth factor receptors. Moreover, JAK-mediated phosphorylation may create docking sites on the receptors for binding of SH2-containing proteins such as STATs, Src kinases, and other signaling intermediates (reviewed in Baker et al. 8 ). JAKs or Src kinases, depending on the nature of the STAT that is being activated, then induce tyrosine phosphorylation and activation of STAT proteins (Fig. 3). These observations suggest that 2 independent pathways can mediate STAT activation, one that is dependent on JAKs and the other that is dependent on other tyrosine kinases, such as Src family kinases (SFKs). Recent studies using targeted inhibitors/therapies support this model. SRC inhibition by either shRNA or dasatinib results in JAK-dependent phosphorylation of STAT3. Sustained SRC inhibition (by drugs such as dasatinib) has also been shown to result in compensatory activation of JAK kinase activity and JAK-STAT3 binding that permits proliferation and survival, even in the absence of SRC activity. 67,68
The notion that different STATs might be phosphorylated by other tyrosine kinases under different conditions is also supported by studies with other tyrosine kinases such as v-ABL and BCR-ABL. Examination of the molecular mechanisms associated with v-Abl–mediated transformation shows that B cells transformed by this oncogene exhibit constitutively activated forms of JAK1 and JAK3 as well as STAT-1, -3, and -5. 69,70 Activated JAK1 in these cells was found to be associated with the v-Abl protein. Similarly, the BCR-ABL oncogene constitutively activates STAT-1 and STAT-5 in a variety of hematopoietic cell systems in vitro. 71-74 While some studies have shown that BCR-ABL has very little if any effect on the activation of JAKs, 71-74 others indicate that the oncogene is required for the activation of JAK2 in hematopoietic cells that are transformed by oncogenic variants of ABL. 75,76
These studies highlight the role of JAKs, JAK2, in particular, in the development of hematological malignancies. Recent discoveries of oncogenic JAK2 translocations and activating mutations further demonstrate its role in these diseases.
Chromosomal Translocations Involving the JAK2 Locus
Rearrangements of the JAK2 gene that lead to constitutively activated tyrosine kinase activity with oncogenic properties have been known for more than a decade. These translocations result in a variety of JAK2 chimeric transcripts, and expression of their resultant fusion proteins often leads to the development of leukemias of both myeloid and lymphoid origins (Fig. 5).
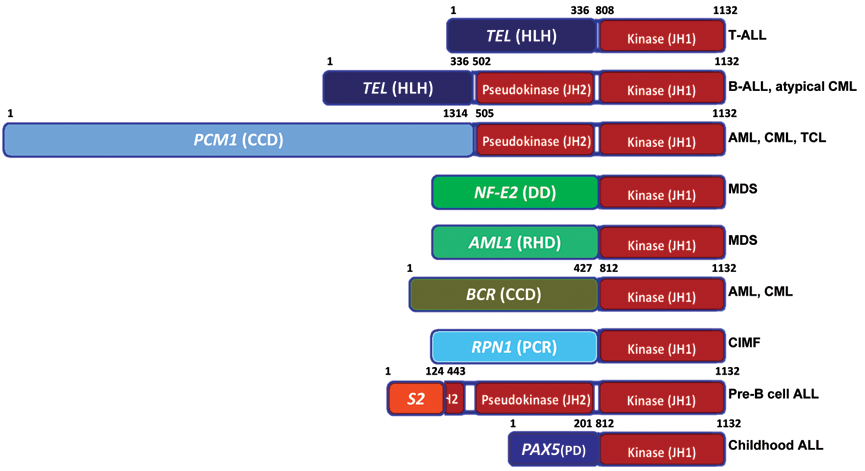
JAK2 translocations discovered in myeloid and lymphoid leukemias. All translocations fuse the multimerization subunit(s) of partner proteins to the JAK2 catalytic kinase (JH1) domain. The amino acid boundaries of each protein in the chimeras are indicated numerically where available, and their associated disease conditions are highlighted. HLH = helix-loop-helix domain; CCD = coiled-coil domain(s); DD = dimerization domain; RHD = Runt homology domain; OLI = oligomerization domain; PCR = proteasome/cyclosome repeated motifs; S2 = SSBP2 fused at LiSH; LiSH = Lis homology motif; PD = paired domain.
ETV6/TEL-JAK2 fusions
The first publication that reported a chromosomal translocation involving the JAK2 gene was published in 1997 and described the t(9;12)(p24;p13) translocation in a case of early pre–B acute lymphoid leukemia. This translocation results in the fusion of the helix-loop-helix (HLH) oligomerization domain of the Ets variant gene 6 (ETV6) with the tyrosine kinase domain (JH1) of JAK2. 77 This initial report was followed by a second study, which describes the now famous TEL-JAK2 fusion that is generated by t(9;12)(p24;p13) in several cases of human leukemia. 78 TEL is a member of the ETS transcription factor family, and the only productive fusions were found to be between its oligomerization domain and the kinase domain of JAK2. The TEL-JAK2 fusion protein was found to have constitutive kinase activity, and its ectopic expression in a murine myeloid cell line resulted in IL3-independent growth. These findings were extensively validated by the induction of growth factor independence, hematopoietic cell transformation, and the development of a lymphomyeloproliferative disease in mice retrovirally transduced with a TEL-JAK2 gene fusion construct. 79 Subsequently, the TEL-JAK2 chimera was shown to activate the PI3′-kinase/AKT and the ERK1/2 signaling pathways. These reports created excitement, as they led to the belief that small molecule inhibitors of JAK2 could be developed to treat leukemias in patients with this specific genetic aberration. However, the scope of such a drug would be limited, given that only a minute fraction of leukemia cases showed the presence of TEL-JAK2 fusions.
PCM1-JAK2 fusions
The discovery of the activating JAK2V617F mutation led to a sudden rise in interest to sequence and genetically analyze the JAK2 locus in patients with hematological indications, and paved the way for the identification of a number of other novel translocations involving the JAK2 gene. The human autoantigen pericentriolar material (PCM1) gene was found to be fused with JAK2 in a German clinical study of male patients afflicted with both acute and chronic leukemia with diverse clinical outcomes. Although these t(8;9)(p22;p24) rearrangements were found to give rise to transcripts with variable breakpoints in both genes, all of the fusion proteins contained a number of the coiled-coil domains (CCD) of PCM1 and the complete catalytic tyrosine kinase domain of JAK2. 80 Two independent French groups isolated similar PCMI-JAK2 translocations in cases of atypical chronic myeloid leukemia and acute erythroid leukemia. 81,82 Subsequently, this genetic aberration was also found in a French study of a patient with T cell lymphoma. 83 While it is known that this translocation leads to constitutive activation of the JAK2 kinase due to the oligomerization mediated by the coiled-coil domains of PCM1, 84 biochemical and in vivo analyses describing the deregulation of the JAK/STAT pathway mediated by this translocation have yet to be reported.
Myelodisplastic syndrome (MDS)–associated translocations
A number of JAK2V617F-negative and Ph-negative patients with chronic myeloproliferative disorder (CMPD) eventually progress to MDS. Cytogenetic studies revealed that most of these patients harbor rearrangements of the JAK2 gene. Novel JAK2-NF-E2 and JAK2-AML1 gene fusions, in addition to previously identified JAK2 rearrangements (e.g., TEL-JAK2), have been identified from such cases. 85 Both NF-E2 and AML1 are transcription factors that contain dimerization motifs. The NFE2-JAK2 fusion is clinically significant because multipotent myeloid progenitors from polycythemia vera (PV) patients express high levels of NF-E2. 86 The case for the JAK2-AML1 fusion is also of interest as AML1 has dozens of other translocation partners, with many of these fusion proteins being causative molecular events in hematological disorders. However, the mechanistic outcomes of these novel JAK2 chimeras and the expression of their predicted fusion proteins have yet to be demonstrated experimentally.
BCR-JAK2 fusions
CML is typified by the Philadelphia chromosome, which leads to the expression of the BCR-ABL1 fusion protein. Detailed cytogenetic analysis of a German patient diagnosed with typical CML led to the discovery of a BCR gene rearranged with JAK2 (instead of ABL1). The t(9;22)(q34;q11.2) translocation was shown to fuse the coiled-coil dimerization domain of BCR with the catalytic JH1 domain of JAK2. Consequently, the patient was unresponsive to imatinib as the drug is a specific ABL1 kinase inhibitor with no inhibitory activity against JAK2. 87 Two years ago, an Italian study reported the presence of t(9;22)(p24;q11) in an acute myeloid leukemia (AML) patient. Although this translocation also leads to the fusion of the BCR and JAK2 genes, the breakpoint in the BCR locus occurs at a different place from that of the German CML patient. 88 Later that same year, an Australian study reported a t(9;22)(p24;q11.2) translocation, leading to a BCR-JAK2 fusion in an atypical CML patient with leukemia cutis. 89
RPN1-JAK2 fusion
The ribophorin 1 gene was found fused to JAK2 due to a unique reciprocal t(3;9)(q21;p24) translocation in an isolated case of chronic idiopathic myelofibrosis (CIMF). 90 While the biochemical consequence of this juxtapositioning is unknown, it is hypothesized that the resultant fusion protein could possess constitutive JAK2 tyrosine kinase activity.
SSBP2-JAK2 fusion
In a recent case of acute pre–B cell lymphocytic leukemia, the t(5;9)(q14.1;p24.1) translocation resulted in the rearrangement of the SSBP2 transcriptional regulator gene with JAK2. 91 Three fusion transcripts were obtained from this clinical study, with each fusing the N-terminal LiSH putative dimerization domain of SSBP2 with exon 11 of JAK2. While a frame shift causes one of these transcripts to prematurely terminate, the other 2 fuse the SSBP2 dimerization domain in frame with the entire C-terminal half of JAK2, which includes both the catalytic and the pseudokinase domains. It is believed that the mechanism for tyrosine kinase activation in this instance is due to the dimerization afforded to JAK2 by the coiled-coil domains of its translocation partners, although supporting evidence at the experimental level has not yet been reported.
PAX5-JAK2 fusion
The most recent fusion partner of JAK2 was discovered last year in a population study involving 446 childhood cases of acute lymphocytic leukemia (ALL), whereby the paired DNA binding domain of the PAX5 transcription factor was shown to be fused to several novel gene partners, including the kinase (JH1) domain of JAK2. 92 The reciprocal rearrangement was also isolated and contained a truncated JAK2 gene that was devoid of its kinase domain fused to the transactivating domain of PAX5. Mechanistically, it is predicted that in the former situation, the leukemogenic effect is mediated by the constitutive activation of JAK2, while in the latter, deregulation of PAX5 transcriptional activity is the causative oncogenic event.
In addition to translocations involving the JAK2 gene, other genetic rearrangements might also lead to its enhanced expression and activity. In a study of Hodgkin lymphoma cell lines, for example, telomeric translocations were found to increase the copy number of several oncogenes, including JAK2. 93
Activating Mutations of the JAK2 Gene
The vast majority of chromosomal translocations of the JAK2 gene lead to leukemias and lymphomas. Similarly, all activating point mutations, deletions, and insertions in this gene lead to myeloproliferative neoplasms that can further develop into leukemias and MDS upon the acquisition of additional genetic lesions (Fig. 6).
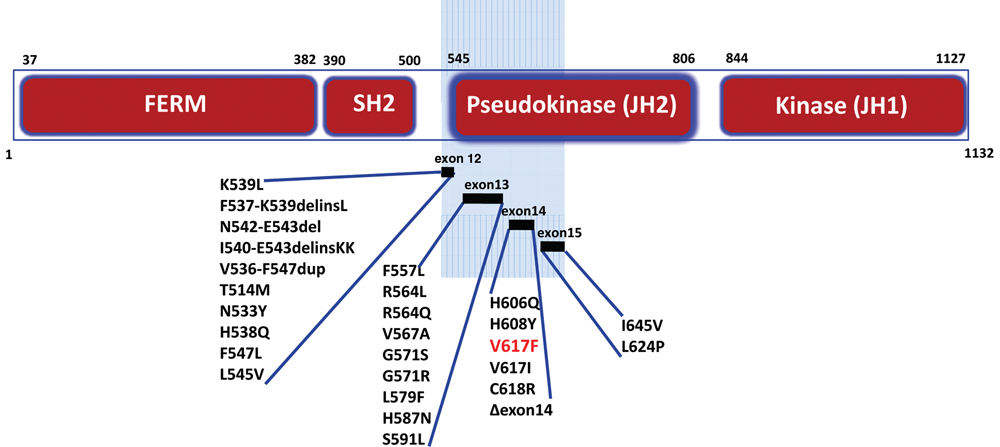
JAK2 mutations found in the clinic. Point mutations, duplications, and insertions identified in MPNs map to exons 12 to 15, revealing the N-terminal half of the regulatory pseudokinase (JH2) domain as a hotspot for kinase activation.
G1849T transversion in exon 14
The clinical relevance of activating point mutations in the JAK family of genes was demonstrated in 2005 when a number of groups analyzing the blood samples from patients with myeloproliferative neoplasms (MPNs) reported the discovery of the V617F mutation borne out of the G1849T transversion at the JAK2 locus. 41-43 This mutation was found in 95% of all PV patients, in half of those suffering from essential thrombocythemia (ET), and in 30% to 50% of those with primary myelofibrosis (PMF). It was soon discovered that this mutation is not unique to BCR-ABL–negative classic MPN, as patients with other myeloid neoplasms also had the JAK2V617F mutation. These neoplasms included approximately 20% to 50% of the cases of refractory anemia with ring sideroblasts and marked thrombosis (RARS-T) and a subset (less than 5%) of cases of acute myeloid leukemia (AML) or MDS. 94 A numerical gain of JAK2 copy number was associated with disease progression in a significant portion of V617F-positive PV patients. 85 Based on the predicted JAK2 structure and atomic level simulations, the V617F substitution is believed to disrupt the autoinhibitory interaction between the pseudokinase (JH2) and kinase (JH1) domains of the kinase. 95 The V617F mutation occurs in a multipotent hematopoietic progenitor cell that gives rise to erythroid and myeloid lineages that are rendered growth factor independent and hence gain a survival advantage over nonmutated cells. 96 These findings were considered so important that within 3 years, the V617F mutation in JAK2 was approved by the World Health Organization (WHO) as the initial diagnostic marker for erythrocytosis, thrombocytosis, and unexplained leukocytosis, splenomegaly, or abdominal vein thrombosis. 97 Patients harboring the V617F mutation, similar to those with the TEL-JAK2 translocation, represent a population that could be treated with specific inhibitors of the JAK/STAT signaling pathway.
Exon 14 deletion and other mutations
Further clinical investigations revealed that exon 14 is deleted irrespective of V617F mutation status in a subset of patients suffering from MPN. These deletions had initially gone undetected because they arise from alternate splicing of the JAK2 transcript and most sequencing efforts had analyzed patient DNA. These patients were found to exhibit the typical symptoms of the disease, much like those who harbor the V617F mutation. More recently, MPN cases have presented with novel mutations of exon 14 including H606Q, H608Y, V617I, and C618R. 98
Exon 12 mutations
As stated above, 95% of PV patients harbor the V617F mutation. Most of the remaining 5% of PV patients were soon found to have other activating mutations in exon 12 of the JAK2 gene. 99 These lesions included the K539L point mutation, the replacement of F537-K539 with a leucine (designated F537-K539delinsL), and the deletion of N542-E543 (N542-E543del). 100 Whereas JAK2V617F mutations are typically homozygous (by mitotic recombination), exon 12 mutations are heterozygous in patients with PV. Because these mutations also arise within the JAK2 pseudokinase domain, it is hypothesized that they interfere with the negative regulation of the catalytic JH1 domain. These exon 12 mutations were found to cause increased JAK2 and ERK1/2 phosphorylation as compared to wild-type or V617F-mutated JAK2. While the morphological changes in the bone marrow of these patients differed, their clinical symptoms were identical to those harboring the V617F mutation. Mutations in exon 12 (as in the case of V617F) have also been included in the diagnostic criteria for CMPD by the WHO. 101 This is particularly significant for Asian populations because the incidence of exon 12 mutations has been shown to be as high as 23% in a study of Taiwanese PV patients. 102 That same study has also uncovered a novel I540-E543delinsKK mutation in exon 12. Other novel exon 12 mutations include the duplication of residues V536-F547 and the T514M, N533Y, H538Q F547L, and L545V point mutations. 98
Exon 13 and exon 15 mutations
Mutations in exons 13 and 15 were discovered in an RNA-based sequencing effort aimed at analyzing the JAK2 locus from peripheral blood samples of approximately 20,000 patients with clinically suspected MPN. 98 These novel mutations included F557L (due to the loss of a single nucleotide causing an early termination at residue 567), R564L, R564Q, V567A, G571S, G571R, L579F, H587N, and S591L in exon 13 and L624P and I645V in exon 15. Like the exon 12 mutations, these mutations also occur in a heterozygous manner.
All of the clinically relevant point mutations and deletions of JAK2 reside in exons 12 through 15 and, hence, occur within the N-terminal half of the regulatory JH2 pseudokinase domain (Fig. 6). These studies further lend credence to the prediction that perturbations within the pseudokinase domain of Janus kinases can have oncogenic manifestations and highlight the importance of this catalytically inactive domain in regulating cytokine-induced cellular proliferation. While the functional significance of the newly discovered exon 12, 13, 14, and 15 mutations has yet to be determined at the biochemical level, they underscore the importance of sequencing the entire pseudokinase domain of MPN patients who test negative for the V617F and exon 12 mutations that form the current diagnostic criteria.
BCR-ABL/JAKV617F double mutants
In the last few years, several CMPD cases have been reported in which both the BCR-ABL translocation and the JAK2V617F mutation are both present in bone marrow samples. 103-106 These studies have revealed that JAK2V617F mutation–associated CMPD develops predominantly after selective treatment of Ph+ CML with imatinib. Further, the emergence of the BCR-ABL translocation on the background of JAK2V617F CMPD appears to be unrelated to prior myelosuppressive treatment (standard therapy for CMPD). Finally, JAK2V617F mutation seems to precede the acquisition of the Ph chromosome. 107 It has been shown that the kinase activity of JAK2 is required for the stability of the BCR-ABL protein and thus maintenance of the oncogenic signal in CML cells. 76 These findings have raised the possibility of using JAK2 inhibitors alone or in combination with imatinib as potential therapeutics for CML patients, regardless of their JAK2 mutational status.
Other Mechanisms of JAK2 Activation
Activation of the JAK-STAT pathway has also been observed in diseases with defects in proteins that signal upstream of the Janus kinases. One such example is the constitutive activation of JAK2 and STAT1 in cells derived from monosomy 7 MDS patients that arises due to aberrant cytokine receptor signaling. 108,109 Monosomy 7 cells show increased expression of a differentiation-defective GCSFR isoform (IV) that fails to internalize following GCSF binding, as would normally occur for the full-length receptor. This receptor variant is also defective in facilitating phosphorylation of STAT-3, but its ability to signal phosphorylation of STAT-1 and -5 is unimpaired. 109,110 Consequently, the ability of these cells to differentiate is limited, although their proliferation via JAK-2 remains unhindered.
The genetic aberrations in JAK2 reviewed above have opened new avenues for diagnosing and classifying patients with myeloproliferative neoplasms. These findings have also identified activated JAK2 as an attractive molecular target for small molecule inhibitors.
Development of Small Molecule JAK2 Inhibitors
The discovery of genetic lesions leading to the activation of JAK2 kinase activity in leukemia and lymphomas along with the obligatory association of MPN (e.g., PV) with activating JAK2 mutations has generated tremendous enthusiasm for the development of JAK2 inhibitors for the treatment of these hematological indications. Consequently, a large number of chemotypes have been identified that possess Janus kinase inhibitory activity. These molecules range from substrate-competitive inhibitors inspired by the structure of the canonical JAK2 inhibitor, tyrphostin AG490, to ATP-competitive pyridones and pyrimidine analogs 111 (Fig. 7). The majority of these compounds were intentionally developed as JAK2 inhibitors and are designated as class I inhibitors. Class II inhibitors were initially developed as inhibitors of other target kinases and were later found to possess JAK2 inhibitory activity. While patents have been filed for a large number of JAK2 inhibitors by a host of pharmaceutical companies and medical institutions, only a fraction of these inhibitors have entered into clinical trials. The therapeutic outcomes of these compounds are currently being assessed. These drugs have been reviewed extensively since the 51st Annual American Society of Hematology Meeting (ASH 2009), 112-115 and we have summarized and discussed the chemical identity (where available), preclinical findings, and current status in clinical trials of these compounds below.
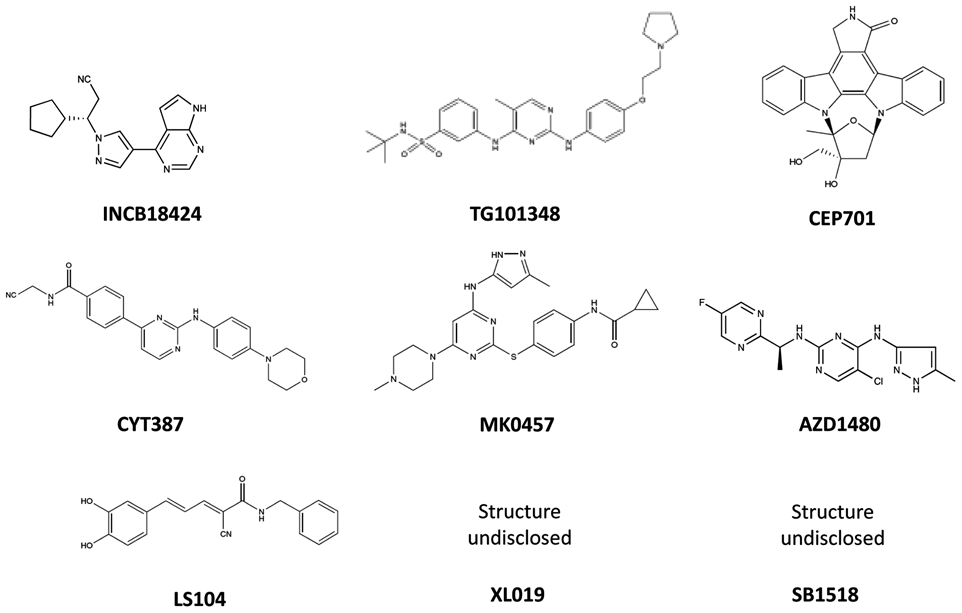
JAK2 inhibitors in clinical trials.
ATP-Competitive JAK2 Inhibitors
Substrate-Competitive Inhibitors of JAK2
Concluding Remarks
Whereas the preclinical results with JAK2 inhibitors for MPN therapy have been promising, these agents have not met with the same degree of success in the clinic. It is now widely accepted that JAK2 inhibitors as single oral agents significantly improve the quality of life of MPN patients by the palliation of debilitating disease-related symptoms such as palpable splenomegaly, pruritis, weight loss, early satiety, and erythrocytosis. This property makes them attractive options for patients with MPN either as primary or second-line therapies in those who have failed hydroxyurea and pegylated interferon. However, to date, these agents do not significantly effect bone marrow fibrosis, alter marrow histopathology, reverse cytopenias, reduce red cell transfusion requirements, or significantly reduce allele burden. Although some patients may have a reduction in allele burden, there are no data to suggest that these agents can eradicate the JAK2V617F-bearing clone. This is in contrast to studies demonstrating that treatment with pegylated interferon can eradicate the JAK2V617F clone and lead to molecular complete remissions and restore polyclonal hematopoiesis in a small percentage of patients with MPN. 119 No phase III study has been completed, and thus, there is no evidence to suggest that these agents can alter the natural history of MPN in the clinic. These agents may palliate disease-related symptoms, but there is no evidence for a curative potential. While the safety profile of these JAK2 inhibitors is acceptable, their associated toxicities of anemia, thrombocytopenia, and gastrointestinal discomfort, although manageable, will require the prudent choosing of candidates. It is certain that JAK2 inhibitors will continue to play an important role in alleviating the debilitating symptoms associated with MPN. Future clinical trials will need to focus on combination targeted therapy as the nature and significance of the MPN stem cell population are further demonstrated at the clinical and molecular levels. 120
Footnotes
EP Reddy is a stockholder, board member, grant recipient, and consultant for Onconova Therapeutics, Inc. SJ Baker is a consultant for Onconova Therapeutics, Inc. Dr. Silverman is a grant recipient of Onconova Therapeutics Inc. Dr. Jatiani declares no potential conflicts of interest.
This work was supported by the Department of Defense [grant number W81XWH-06-1-0267]; the National Heart, Lung, and Blood Institute [grant number HL080666]; and Onconova Therapeutics Inc.