
Abstract
p97 (also called VCP in metazoans and CDC48 in yeast) is a highly conserved, abundant, and essential type II AAA ATPase that functions in numerous ubiquitin signaling–dependent processes. p97/Cd48 activities require a growing number of adaptor or accessory proteins that promote interactions with ubiquitinated proteins. p97 has human disease relevance as it is mutated in familial cases of inclusion body myopathy associated with Paget disease of the bone and frontotemporal dementia (IBMPFD). There is also increasing evidence suggesting that p97 and/or some of its adaptors play a role in cancer. This review will summarize our existing knowledge of the biochemical, molecular, and cellular activities of p97-containing complexes, with an ending focus on their potential role in malignancy.
Overview of Ubiquitin Signaling
Ubiquitination is a highly conserved and essential posttranslational protein modification process that regulates the expression and/or activity of numerous proteins in the eukaryotic cell (reviewed in Hochstrasser 1 and Hershko and Ciechanover 2 ). In this process, ubiquitin, a small 76–amino acid protein, is covalently attached to lysine residues of a substrate through the cooperative activity of ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin ligase (E3) enzymes. Proteins can be modified by a single ubiquitin moiety, but more commonly, multiubiquitin chains form on substrates via E3-driven attachment of ubiquitin on internal residues of ubiquitin itself. Ubiquitin harbors 7 lysine residues, all of which may be used for chain formation. 3 Thus, the structure of the ubiquitin chain can be diverse and have differing consequences on the fate of the modified substrate.
The ubiquitin signal derived from a lysine-linked 48 ubiquitin chain is the best studied. Substrates harboring these chains are targeted for degradation by the proteasome (reviewed in Finley 4 ). Another very common ubiquitin modification is chains carrying lysine 63 linkages (reviewed in Chen and Sun 5 ). This type of chain induces lysosomal-mediated degradation of many cell surface proteins including receptors, channel proteins, and permeases and may target misfolded proteins to the aggresome-autophagy degradation pathway. It also modulates protein function independent of promoting degradation, such as by activating kinases and DNA repair proteins. While many ubiquitin-signaling events rely on the formation of ubiquitin chains, modification of substrates with a single ubiquitin moiety can also have important functional consequences. It has been shown that monoubiquitination of receptors in mammalian cells is sufficient for promoting their internalization (reviewed in Acconcia et al. 6 ).
Ubiquitination serves as a signal to promote new interactions that are mediated by proteins that harbor ubiquitin association domains or interaction motifs (reviewed in Hicke et al. 7 ). These interactions are critical for mediating the fate of the ubiquitin-modified protein. There are numerous types of ubiquitin association domains, and it is estimated that there are close to 300 ubiquitin-binding domain–containing proteins in humans. Ubiquitin-binding proteins can function on their own or in the case of p97 act as adaptors or accessory proteins that recruit the enzyme to specific substrates (reviewed in Elsasser and Finley 8 and Schuberth and Buchberger 9 ).
Biochemistry of p97-Containing Complexes
p97 Structure
Considering the cancer relevance of this review, I will generally refer to the ATPase as p97, unless I refer to functions specific for the yeast protein. p97 is a member of the type II AAA (
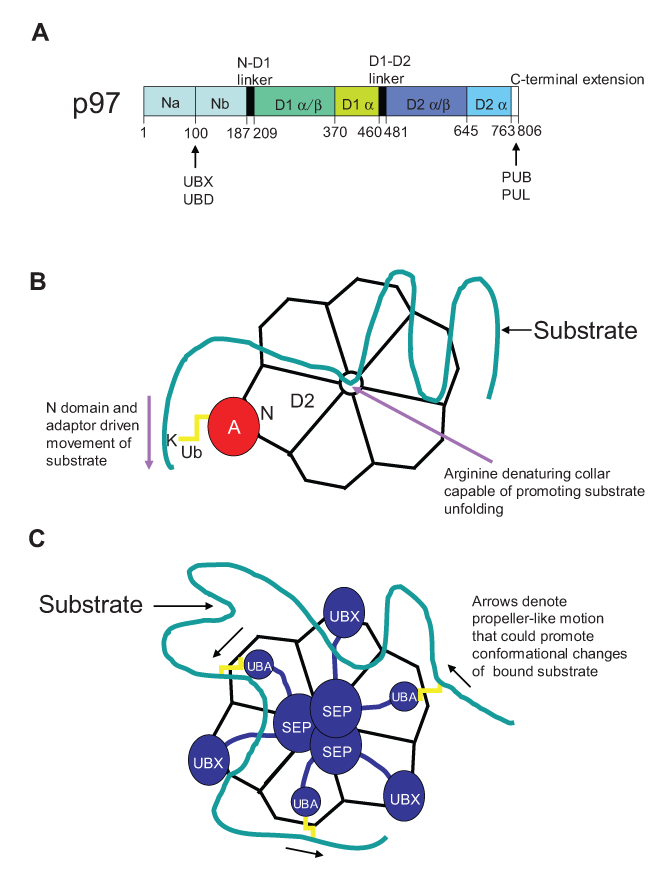
Structure and function of p97. The modular structure of the enzyme is depicted (
There are a number of proposed models (Fig. 1B and 1C) based on structural studies by which p97’s nucleotide-binding and hydrolysis function results in the change of p97 and/or adaptor conformation and results in the application of mechanical movement or force on substrates. In one model, ATP hydrolysis by the D2 domain transmits conformational changes in the hexamer that spreads through the D1-D2 linker and leads to the movement of the amino terminal adaptor–binding domain, which is presumably bound to a substrate. 13 Repeated cycles of ATP hydrolysis and movement of the substrate-bound amino terminal domain may provide a ratcheting effect on substrates and facilitate their movement, a process likely important for the extraction of misfolded proteins from the endoplasmic reticulum (ER). The p97 hexamer also contains ATP hydrolysis–dependent moveable arginine and hydrophobic ring loops in the pore at the D2 end. The hydrophobic ring has been proposed to bind substrates, while the “arginine denaturation collar” has been postulated to promote substrate unfolding or maintain the unfolded state. 15 These arginine residues are required for p97 function in ERAD (ER-associated degradation). 15 Structural studies with p97 in complex with adaptor p47 also suggest that D2-dependent ATP hydrolysis leads to conformation changes that can spread through the adaptor. 16 In the case here in which the stoichiometry is 1 p97 hexamer per 3 adaptors, this could lead to a propeller-like motion and promote conformation changes of p47-bound substrates.
There are other notable characteristics of p97’s structure that deserve mentioning. First, in contrast to D2, D1 prefers ADP and does not hydrolyze ATP under physiological conditions, 17,18 is stable, 19 and may play a role in p97 hexamerization. 20 In addition, while the hexamer has a central pore, it is blocked by a Zn++ ion that is coordinated by histidine residues facing inwards towards the channel in the D1 region. 13 Thus, it is unlikely that proteins pass through the central pore of p97. Notwithstanding, these models are based on structures of p97 or p97 with one adaptor so it remains unclear how they will apply to the diverse array of p97 complexes or their substrates. Structural studies with p97 adaptor-substrate complexes are obviously needed. However, this will be no easy task considering that many substrates are either membrane bound or misfolded and aggregate prone.
p97 Accessory Proteins
While p97 functions on ubiquitin-modified proteins, it does not harbor a high-affinity ubiquitin interaction domain. The ubiquitin-binding activity of p97 is mediated by a growing number of adaptor or accessory proteins, which harbor different types of p97 interaction motifs and in many cases ubiquitin association domains (reviewed in Elsasser and Finley,
8
Schuberth and Buchberger,
9
and Yeung et al.
21
). The greatest numbers are members of the UBX domain–containing family of adaptors (UBXD1, UBXD2, UBXD3, UBXD4, UBXD5, UBXD7, UBXD8, FAF1, SAKS1, ASPL, p37, p47, VCIP135, and YOD1) (reviewed in Schuberth and Buchberger
9
). This domain adopts an ubiquitin-like β-grasp fold.
22
The well-studied adaptor Npl4 harbors a similar motif called a UBD (ubiquitin D) domain and binds to the same amino terminal region of p97.
23,24
Unlike other UBX domain–containing proteins, its interaction with p97 requires binding to another protein called Ufd1 (it too harbors a p97-binding domain), which allows for an interaction of the dimeric complex with p97.
23,24
A motif termed the PUG or PUB (I will refer to it as PUB) domain also serves as a p97-binding domain
25
and includes PNGase, RING finger 31, and UBXD1. Curiously, while UBXD1 has 2 putative p97 interaction motifs, recent studies indicate that the PUB, and not the UBX, domain mediates binding to p97.
26,27
The inability of the UBX domain of UBXD1 to bind p97 may be due to the absence of conserved hydrophobic and proline residues in a region (located between β strands 3 and 4) responsible for p97 contact. The role that the UBX domain plays in UBXD1 function remains to be defined. An important distinction between the UBX and PUB motifs is that they contact different regions of p97. The UBX and UBD motifs bind the amino terminal domain, while the PUB domain associates with the carboxy terminal tail.
23,26,27
While this may mean that a p97 complex simultaneously binds the UBX/UBD and PUB domain–containing adaptors, this may not always be the case. In fact, UBXD1 has been shown to compete for p47 binding to p97.
27
This is due to UBXD1 harboring another p97-binding region within its amino terminus that interacts with the amino terminal of p97.
27
The PUL domain (present in human
The function of p97 accessory proteins has been classified generally as substrate-recruiting or -processing factors (reviewed in Jentsch and Rumpf 30 ). Substrate-recruiting factors harbor ubiquitin association domains and do not possess enzymatic activities, although they may certainly interact with others that do. Substrate-processing factors possess enzymatic activity (and in many cases, ubiquitin-binding domains as well) and can modify substrates by ubiquitination (e.g., E4B/Ufd2 and perhaps RING finger 31), deubiquitination (e.g., YOD1, VCIP135, and Ataxin-3), and in the case of PNGase, degylcosylation. There is a strong likelihood that many adaptors will have dual functions in substrate-recruiting and modulating posttranslational modification, either by themselves or via associating partners.
p97 Adaptor Stoichiometry
Considering that p97 exists as a hexamer and each monomer harbors at least 2 adaptor-binding domains, one can envision the formation of complex macromolecular structures. The stoichiometry of p97 adaptor binding appears to be adaptor dependent. In the case of Npl4-Ufd1, the stoichiometry is one adaptor per one hexamer. 31 For p47, however, it is 3 molecules per p97 hexamer. 16 It has also been shown that Cdc48 can bind multiple adaptors simultaneously as in UBX4 and Npl4-Ufd1 32 or associate with numerous adaptors that function in a given degradation pathway as in ERAD. 32-34 In this case, Cdc48 binds UBX2 (a recruiter of Cdc48 to the ER membrane), Npl4-Ufd1 (mediates an interaction with misfolded ubiquitinated ER resident proteins), and UBX4 (a protein required for ERAD whose biochemical function remains to be defined). 32-34 The point here is that the makeup of the p97-containing complex is likely multifaceted and comprised of numerous adaptors that supply critical cooperative functions and/or interchangeable ones that provide sequential activities to specific biochemical processes.
Function of Nonubiquitin-Binding Domain p97 Adaptors and Mechanisms Conferring Specificity to p97 Complex-Substrate Interactions
As stated above, the largest family of p97 adaptors is those that harbor a UBX domain. Interestingly, the majority of these adaptors (8/14 in humans) do not harbor known ubiquitin association domains and do not appear to interact, at least directly, with ubiquitinated substrates. 35 Future studies are obviously needed to elucidate their biochemical and molecular activities. Do they function as competitive inhibitors for those adaptors that harbor ubiquitin interaction motifs? Do they function in recruiting other proteins to p97-containing complexes that are substrate bound? Do they even promote ubiquitin-independent activities of p97?
Another big question that remains in terms of p97 adaptors is what dictates specificity of adaptor-substrate interactions beyond association with the ubiquitin tag. If p97-containing complexes regulate a plethora of substrates (this seems possible considering its abundance), is the limited number of adaptors identified to date sufficient to explain the targeting of a large number of molecules? Are we missing other families of p97 adaptors? If not (which is likely considering the results of thorough proteomics-based interaction studies), 35 specificity may be dictated by the combined binding affinities of different adaptors or accessory proteins, each of which have their own binding preferences for different regions on a target. It is also conceivable that p97 does not regulate a diverse array of proteins but those of higher abundance that have common characteristics, such as ubiquitinated misfolded substrates. It will certainly be of interest to determine if p97-containing complexes regulate a large number of properly folded proteins and, if so, determine mechanisms governing substrate recognition specificity.
Activities of p97-Containing Complexes
Delivery of Misfolded ER-Localized Proteins to the Proteasome
p97/Cdc48 activities on substrates can be divided into 3 general categories: promoting substrate degradation, facilitating segregation of ubiquitin-modified proteins from other interacting nonmodified molecules, and modulating substrate ubiquitination in both a positive and negative manner. The first part of this section will cover its role in the targeting of substrates to various degradation pathways (Fig. 2).
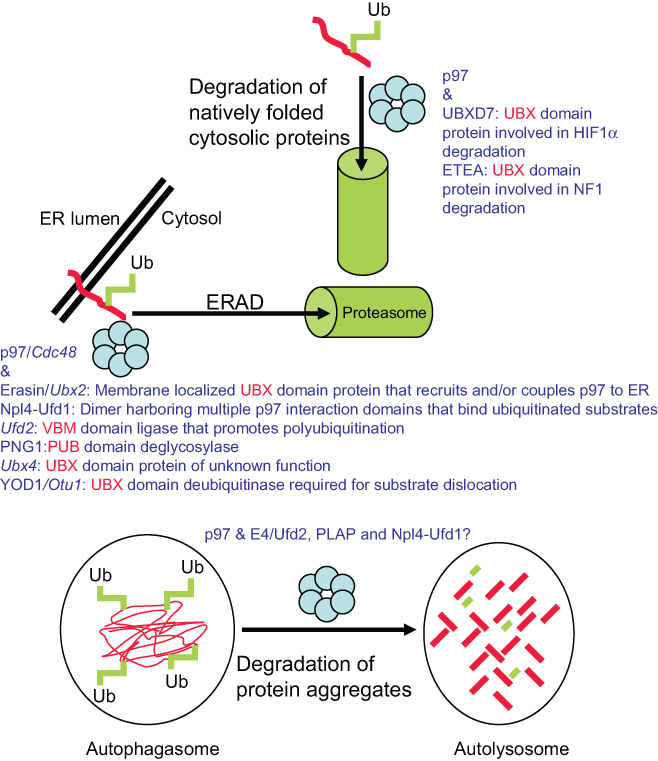
p97 participates in various protein degradation pathways. The role of p97 and adaptors in ubiquitin signal–dependent degradation of natively folded cytosolic proteins, misfolded ER-localized proteins, and aggregate-prone cytosolic proteins.
The functional contribution of p97/Cdc48 and various adaptors has been defined best in the ERAD pathway and reviewed previously. 36,37 Briefly, ERAD is a highly conserved process that is responsible for the clearance of misfolded proteins that are localized at or in the ER. Protein domain misfolding can occur in the ER lumen, the ER membrane, and even in a cytosolic-facing region for an ER-anchored protein. For those proteins that cannot be refolded by the various chaperone systems, they are delivered to the ubiquitination machinery, ubiquitinated, retrotranslocated from the ER, and degraded by cytosolic proteasomes. One important function of p97 is to promote retrotranslocation of misfolded ubiquitinated substrates. As discussed above, p97’s enzymatic activity is required to maintain protein misfolding (and perhaps even further denaturation) and the movement of the misfolded protein through the ER channel. Degradation of misfolded ER-localized proteins also requires the p97/Cdc48 adaptor Erasin/Ubx2 (like p97/Cdc48, I will refer to the mammalian name first and the budding yeast name second if different), which is an ER-spanning protein that anchors the ATPase to the membrane. 33,34,38 In addition to Ubx2, other p97 adaptors play important roles in this process. These include Npl4-Ufd1 (couples p97/Cdc48 to ubiquitin-modified substrates via its ubiquitin-binding function), 39 Png1 (deglycosylates misfolded glycoproteins, although the yeast homolog does not harbor a PUB or other Cdc48 interaction motif), 40 E4B/Ufd2 (ligase responsible for further substrate polyubiquitination and enhanced proteasome delivery), 41 YOD1/Otu1 (a deubiquitinating enzyme, whose required function is required for substrate translocation), 42 and Ubx4 (a nonubiquitin association domain containing adaptor in yeast, whose function remains undefined). 32 Although there is now good information pertaining to the number, types, and activities of adaptors involved in ERAD, there are still many unresolved issues. Do these adaptors function in the same hexameric complex, or do they sequentially act and cycle on and off the hexamer? If they cycle on and off (this is likely with those adaptors that bind to a common site on p97 such as the UBX domain–containing proteins), what are the mechanisms that control cycling? Do adapter-driven activities such as ubiquitination and deubiquitination require the ratcheting or unfolding activities of p97, or does it function simply as a scaffold in these processes? There is still much to learn about p97-containing complexes in the destruction of misfolded ER-localized proteins and even natively folded proteins that are ER localized and degraded by this pathway.
Promoting the Degradation of Aggregate-Prone Proteins by the Aggresome-Autophagy Pathway
p97 also participates in the degradation of misfolded aggregate-prone cytosolic proteins by the aggresome-autophagy degradation pathway. 43-46 This pathway is responsible for degrading misfolded proteins that accumulate and aggregate in the cytosol. Aggregate proteins are first ubiquitinated by the E3 ligase Parkin, and this ligase appears to prefer building lysine 63–linked chains on substrates (reviewed in Chin et al. 47 ). This ubiquitin signal promotes an interaction with ubiquitin association domain–containing protein HDAC6 and through cooperative interactions with HDAC6 and the dynein motor–complex retrograde transport of the misfolded proteins towards the microtubule-organizing center. 48 The center is located outside the nucleus, and the continued depositing of ubiquitinated misfolded proteins leads to the formation of a distinct ubiquitin-staining positive structure called the aggresome. p62 (also harbors an ubiquitin association domain) facilitates the recruitment of autophagic membranes to the aggresome to form the autophagosome (reviewed in Bjorkoy et al. 49 ). The subsequent fusion of this structure with the lysosome (forms the autolysosome) results in the degradation of misfolded aggregate proteins. Interestingly, recent studies indicate that HDAC6 is essential for this process in murine fibroblasts and is involved in recruiting an F-actin network to promote autophagosome-lysosome fusion. 50
There is now excellent genetic, cellular, and animal data implicating p97 in this process. Recent studies have shown that p97 knock-down results in the accumulation of autophagosomes within the cell. 45 These structures are, however, unable to mature into autolysosomes and degrade protein aggregates contained within these structures. These phenotypes are also observed in cells transfected with p97 mutants (residing in the N-terminal and D1 domains) present in familiar IBMPFD patients. 45,46 Moreover, transgenic mice constitutively expressing the most common IBMPFD p97 mutant (R155H) and a mutant (A232E) found in a more severe form of the human condition have been generated, and impressively, they develop IBMPFD-like disease. 51 The mice display degeneration of tissues affected in human IBMPFD (muscle, brain, and bone) and similar cellular pathologies (accumulation of TDP-43 in the cytoplasm of muscle and brain, rimmed vacuoles in muscle, and increased ubiquitin staining) found in human IBMPFD. A recent report looking at a limited number of adaptors/accessory proteins has provided evidence that IBMPFD mutations promote conformational changes in the amino terminal domain, leading to differential interactions with a subset of accessory proteins such as E4B/Ufd2 and Ataxin-3. 52 Interestingly, even though both proteins harbor the same VBM p97 interaction motif, these mutations suppress a p97 interaction with E4B/Ufd2, while they promote Ataxin-3 binding. 52 While these results raise some interesting questions regarding the binding specificity of different VBM domain–containing proteins, they do provide important evidence for the loss of E4B/Ufd2 function and/or gain of Ataxin-3 activity in IBMPFD disease.
The mechanism by which p97 promotes degradation of aggregate proteins by the aggresome-autophagy pathway still remains unclear. Based on the above-mentioned interaction data with IBMPFD mutants,
52
it is possible that the ubiquitin ligase activity of E4B/Ufd2 is involved and that p97 is required to recruit the ligase to the aggregate-prone material and/or control the structure of misfolded substrates so that they can be ubiquitinated by E4B/Ufd2. There is also evidence that p97 interacts with HDAC6,
53,54
and interestingly, loss of p97 or HDAC6 leads to a defect in autophagosome-lysosome fusion.
45,46,50
Thus, p97 may be involved with HDAC6 in promoting F-actin network formation required for autophagosome-lysosome fusion. It is also tempting to speculate that the PUL domain–containing p97 adaptor PLAP (
Degradation of Soluble Proteins by the Proteasome
The adaptor Ufd1 was initially identified in yeast as a gene required for proteasome-dependent degradation of a short-lived, soluble ubiquitin-fused β-galactosidase model substrate (termed UFD substrate). 56 Since this discovery, it is now known that p97/Cdc48 and/or its adaptors play a role in degrading a number of presumably properly folded natural substrates in a variety of organisms such as protein kinase Cdc5 57 and Far1p 58 in yeast and IkBα, 59 HIF1α, 35 and neurofibromin-1 (NF1) 60 in human cells. It remains unclear, however, how p97/Cdc48-containing complexes function in the degradation of these substrates. Does it function simply as scaffold by coupling ubiquitin ligases so that they can provide the ubiquitin degradation tag? Does it segregate the ubiquitin-modified substrate from any interacting partners that may hinder proteasome-dependent degradation? Is there a p97/Cdc48 substrate–unfolding process that is required before certain proteins can be recognized or degraded by the proteasome? A recent report using Drosophila and UFD model substrates has provided strong evidence that p97 and its ATPase activity are required for substrate unfolding and that this process is required for proteasome-dependent degradation. 61 Interestingly, adding a flexible extension to the carboxy terminus of this model substrate bypasses the need for p97, indicating that p97’s denaturing activity may be required for the degradation of those proteins that are structurally resistant for proteasome recognition and/or degradation. This study also documented that while Npl4-Ufd1 was required for the degradation of the model substrate, RNAi-mediated knock-down of individual UBX domain proteins had little or no effect. Although one cannot rule out functional redundancy, these results indicate that unlike with ERAD, UBX-containing adaptors do not provide a common activity required for proteasome-dependent degradation of natively folded cytosolic proteins. Considering that the p97 adaptors UBXD7 35 and ETEA 60 have been shown to promote degradation of specific substrates, perhaps they function more as specificity factors in p97 targeting.
Segregation, Not Degradation
p97’s ortholog in yeast, Cdc48, was originally proposed to have ATP-dependent segregase activity based on its ability to separate a membrane-tethered transcription factor from its membrane-bound binding partner. 62 It was later shown in vitro that this process is dependent on polyubiquitination of the membrane-bound anchor and the dimeric adaptor complex made up of Npl4-Ufd1. 63 Interestingly, in this case, the polyubiquitinated product is not targeted to the proteasome for degradation. This ubiquitin-dependent–proteasome-independent segregase activity of the ATPase-Npl4/Ufd1 complex also plays an important role in the extraction of ubiquitinated Aurora B kinase from chromatin during the exit of cells from mitosis. 64 In addition, it has recently been shown that this activity is important in yeast for the removal of ubiquitinated transcriptional repressor complex from promoter elements. 65 Again, this Cdc48-dependent segregation/extraction process does not promote substrate degradation. Like many p97/Cdc48-dependent processes, we still have only a very basic understanding of its segregase function. Does this segregase activity apply only to protein complexes in which the ubiquitinated protein is not targeted to the proteasome? How is the ubiquitin signal removed after completion of the segregase reaction? Presumably, p97/Cdc48-associated deubiquitinating enzymes perform this function, but this has yet to be shown. Do these substrates carry an ubiquitin tag that does not promote proteasome delivery, or is the ubiquitin tag removed after segregation? Also, does p97/Cdc48 promote global protein unfolding, or does it act on a specific domain(s) of a substrate in complex disassembly reactions? Finally, how universal is this ubiquitin-dependent/proteasome-irrelevant segregase activity of p97?
One topic that I will not cover in much detail in this review is the role of p97-containing complexes in homotypic membrane fusion events. This is one of the original described functions of the ATPase, even prior to our knowledge that it works on ubiquitinated proteins. 66 Although we still have a poor understanding of the role that ubiquitin plays in the process, it appears that at least during the process of Golgi reassembly at the end of mitosis, there is a p97- (mediated by the UBX and UBA domain–containing adaptor p47) and proteasome-independent activity required for membrane fusion. 67 It has been speculated that this may be dependent on the segregase function of p97, 68 although the ubiquitin-modified substrate and binding partners subject to this activity have yet to be identified. A p97-p37 adaptor (harbors a UBX domain, but not an ubiquitin association domain) complex has been shown to function in ER and Golgi fusion events in interphase and at the end of mitosis. 69 This complex appears to work on a different set of fusion mediators 69 (p115-GM 130 tethering and SNARE GS15 rather than syntaxin 5), and it will be interesting to determine in the future if these and other membrane fusion processes are dependent on the segregase activity of p97.
Modulation of Substrate Ubiquitination
p97 has been shown to interact, both directly and indirectly, with numerous ubiquitin ligases and deubiquitinating enzymes (also termed substrate- processing factors), raising the possibility that it may function both positively and negatively in regulating ubiquitin chain formation on substrates and ultimately play opposing roles in modulating their expression or function. 30 Because of these activities, p97/Cdc48 has been termed a molecular gearbox. 30 While it is clear that p97/Cdc48 can interact with both ligases and deubiquitinating enzymes, there is still scant evidence that it directs these different activities to the same substrate. In fact, there is evidence that these p97-regulated opposing activities are required for the same biochemical outcome. For example, the p97-binding factor and deubiquitinating enzyme Ataxin-3 is required for ERAD, and it has been postulated that its chain length–editing function is important for release of the ubiquitinated from the membrane and delivery to the proteasome. 70 Also, a VCIP135 (a UBX domain–containing deubiquitinating enzyme) containing p97-p47 complex is required for membrane fusion at the end of mitosis. 71 As an upstream ubiquitin modification occurring during membrane fragmentation is also required for this process, 71 it will be interesting to see if it is the same substrate that is modified by a p97-ligase complex, then unmodified subsequently by the VCIP135-containing one. Much work lies ahead in determining the biochemical role of p97/Cdc48 in modulating substrate ubiquitination and the biological consequences of these activities.
Posttranscription Regulation of p97-Containing Complexes
A number of phosphorylation sites on p97 have been identified, and a few of these have been demonstrated to have functional significance. p97 is phosphorylated at 2 tyrosine residues on its carboxy terminus, Tyr 796 and Tyr 805 (the second to last amino acid in the protein), upon T-cell antigen receptor binding and T-cell activation. 72 Tyr 805 can also be phosphorylated by c-Src. 73 Interestingly, Tyr residue 805 is required for binding to the PUB domain of PNGase and PUL domain of PLAP, and phosphorylation of this domain abrogates binding to both of these adaptors. 73 It is therefore likely that signal/kinase-induced Tyr 805 phosphorylation will inhibit these adaptor-p97 interactions in cells and modulate p97 complex activity. Consistent with this idea, c-Src has been shown to suppress degradation of a model ERAD substrate and promote the accumulation of ubiquitinated proteins. 73 It remains unclear, however, if this is due to disruption of p97-PNGase or p97-PLAP interactions. Similarly, it is unknown if these associations are affected during T-cell activation. The phosphorylation of Tyr 805 is also modulated in a positive manner by Jak-2 kinase and in a negative manner by protein-tyrosine phosphatase PTPH1. 74 Investigators have shown using a cell free system that Jak-2–mediated phosphorylation promotes p97-p47 complex disassociation from T-SNARE syntaxin 5 and membranes and inhibits transitional ER assembly, while PTPH1 activity has the opposite effect. 74 Again, it remains uncertain how Tyr 805 phosphorylation may modulate this p97-dependent function, although it may modulate binding of an adaptor to the carboxy terminus of p97 that is involved in this process.
Phosphorylation of p97/Cdc48 has also been implicated in changing its subcellular localization during the cell cycle. In yeast, Cdc48 enters the nucleus in the early G1 phase of the cell cycle, while in cultured human cells, p97 localizes to centrosomes during mitosis. 75 Phosphorylation of the penultimate carboxy terminus tyrosine residue of Cdc48 has been linked to its nuclear mobilization. 75 In contrast to the above-discussed mechanism in which phosphorylation modulates adaptor binding, tyrosine phosphorylation of this carboxy terminal residue has been proposed to induce a conformation change in Cdc48 structure, unmasking a nuclear localization signal. 75 Whether such a mechanism exists in regulating nuclear targeting of mammalian p97 remains unclear. Also, the kinase responsible for phosphorylating Cdc48 and regulating this localization has yet to be identified.
The prosurvival serine/threonine kinase AKT has also been shown to phosphorylate p97. p97 has 3 consensus AKT sites (Ser 351, Ser 745, and Ser 747), and all 3 sites appear to be targeted by the kinase in cells. 76,77 The influence of AKT-induced p97 phosphorylation on the functions of the AAA ATPase has yet to be defined. There is one report that it may inhibit its ability to bind ubiquitinated proteins, 76 but it is currently difficult based on the location of these sites and p97 structure and function to evoke a mechanism for this effect. Also, while it has been linked to a prosurvival and NF-κB activation function of p97, 77 it remains to be seen whether there is a direct link between these activities.
p97 phosphorylation has been documented to occur during a number of different experimental conditions and in a variety of cell types (reviewed in Ewens et al. 78 ). While it has been easy to identify p97 as a phosphoprotein and even pinpoint sites of phosphorylation (presumable because it is a very abundant protein), much work lies ahead to understand the functional significance of these modifications. It is likely that phosphorylation and other posttranscription modifications (there is also a report the p97 can be acetylated 79 ) of p97 and its binding partners are critical in controlling interactions and functions, and we need a much better understanding of these processes. We already know that the p97 adaptor p47 is regulated by phosphorylation during the cell cycle, and this posttranslational modification during the early parts of mitosis inhibits binding to Golgi membranes. 80 This phosphate group is then removed at the end of mitosis, allowing p47 with its collaborators p97 and VCIP135 to promote Golgi reformation. 80 It will not be surprising to find that the substrate-binding function of other p97 adaptors is regulated by signal-specific phosphorylation.
Cancer Relevance of p97-Containing Complexes
p97 has been shown to possess numerous activities and participate in many biological processes, some of which are important for cell homeostasis. Not surprisingly, perhaps, the ATPase is essential for the life of nontransformed and cancer cells. 81,82 Does this mean that its relevance to cancer cell proliferation will just parallel its essential roles in normal cells? What evidence do we have that p97 or its adaptors play important roles in proliferation, differentiation, cell survival, and/or maintaining genomic integrity pathways and that alterations in p97-regulated functions are selected during the evolution of cancer cells and are important for maintaining tumor cell proliferation or viability? As discussed above, p97 and/or adaptors have been implicated in the direct regulation of some key cancer-relevant proteins, such as promoting the degradation of and/or negatively regulating HIF1α (tumor angiogenesis and metastases promoter), 35 IκBα (a potential inhibitor of the prosurvival functions of NF-κB), 59 Aurora B kinase (overexpressed in cancer cells and implicated in genomic instability), 64 and NF1 (tumor suppressor and inhibitor of Ras signaling) 60 (Fig. 3). Also, elevated levels of p97 have been reported in a number of human malignancies, including being more prevalent in aggressive and poor outcome cases. 83-91 Based on this expression data, it is possible that elevated levels of p97 contribute to the malignant process through its ability to down-regulate or inhibit growth inhibitory proteins. However, considering that p97’s targets are likely to comprise both growth inhibitory and promoting proteins, this simple mechanism of action is unlikely. Even if we were to assume that IκBα is the predominant p97 target in cancer cells, it is hard to envision how increased expression of an already highly abundant protein, which is likely in excess of other adaptors and cofactors involved in IκBα degradation, enhances IκBα degradation. An alternative explanation for the p97 expression data is that the protein is up-regulated by protein damage-induced stress signals that are elevated in cancer cells. The resulting increase in p97 expression helps in the clearance of abundant, misfolded/aggregate-prone, and potentially toxic proteins from malignant cells and facilitates their survival.
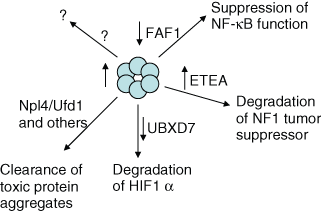
p97 in malignancy. Potential functional roles of p97 and adaptors in cancer cells are depicted. An upward arrow designates how deregulation of the regulated process by overexpression may promote the proliferation or survival of cancer cells, while a downward arrow designates how underexpression may facilitate malignancy.
If genetic mutations are to inhibit p97 function in cancer cells, they are likely to alter specific adaptors. In fact, there is good evidence that at least 2 p97 adaptors, FAF1 and ASPL, display altered expression in tumor cells. Haploinsufficiency of the FAF1 gene in primary tumors has been shown in 30% of uterine cervix carcinomas 92 and in 12.5% of mantle cell lymphomas. 93 FAF1 has also been shown to be down-regulated at the protein level in gastric cancers 94 and a very high percentage of malignant mesothelioma cell lines and primary tumors. 95 FAF1 haploinsufficiency has been observed in a high percentage of tumors arising in a mouse model of the disease (asbestos-treated mice harboring one copy of the p19ARF tumor suppressor). 95 Based on these findings, it will be interesting to see if FAF1 −/+ mice (FAF1-null mice are not viable) are susceptible to tumor formation. Nevertheless, these genetics studies point to a tumor suppressor function for FAF1. In terms of FAF1 activity, the protein has been shown in overexpression experiments to suppress degradation of ubiquitinated substrates, including IκBα. 96 The ability to suppress IκBα degradation and consequently inhibit the growth-promoting/cell survival functions of NF-κB via cytoplasmic sequesterization may represent an important tumor suppressor function of FAF1. However, there are many unknowns about this potential activity that need to be resolved. First, p97 has been proposed to have an opposite effect on IκBα degradation (i.e., promoting it). 59 There is currently very little evidence that FAF1-induced IκBα stabilization is mediated by p97, so it is possible that this activity is p97 independent and a different p97 adaptor promotes degradation. Interestingly, a recent report has shown that FAF1 inhibits IκB kinase activation by disrupting kinase complex assembly. 97 When taking into account this mechanism, down-regulation of FAF1 would also accelerate IκBα degradation and lead to an increase in NF-κB function. However, it remains unclear if the ubiquitin-binding and UBX domains of FAF1 are required for this activity or if this function involves p97. While the above data certainly point to a role for a p97-FAF1 complex in NF-κB pathway regulation, there are mechanistic details that still need to be worked out.
ASPL or TUG is a member of the nonubiquitin-binding domain–containing family of UBX domain–containing adaptors. Its expression is clearly altered in a rare and unusual cancer, alveolar soft part sarcoma (represents 1% of soft tissue sarcomas), through a translocation between chromosomes X and 17 in all cases. Eighty percent of these translocations are nonreciprocal in nature in which gains in Xp11-pter and loss of 17q25-qter occur. 98 A reciprocal translocation between these chromosomes has also been observed in a subset of rare renal cell carcinomas that arise in children. 99 This translocation is 1 of 6 that have been characterized to date in which a region from another chromosome is fused to the TFE3 gene. 100 Those translocations involving ASPL result in the replacement of the amino terminal region of transcription factor TFE3 with the amino terminal half of ASPL. Two types of TFE3 translocation have been described, depending on where in the TFE3 gene translocation occurs. In type I, the transcription factor retains its nuclear localization signal, basic helix-loop-helix DNA-binding domain, and leucine zipper dimerization but lacks its acidic domain, while in type II, the fusion retains all of its known functional domains, including the acidic domain. The ASPL component of these fusions contains the amino terminal half of ASPL, which harbors an ubiquitin-like domain, but not the UBX domain. While it is highly likely that these fusions lead to deregulated TFE3 function (could be a loss or gain of function alteration), it remains unclear how the addition of ASPL sequences influences TFE3 activity. It is also possible that removal of the amino terminal domain of ASPL affects p97-dependent and -independent functions (which have yet to be identified, although there is one report that it regulates the GLUT4 glucose transporter) 101 and contributes to the malignant process.
Although there are no genetic or expression data to support deregulation in cancer, there are 2 adaptors, UBXD7 and UBXD8/ETEA, that at least based on recent functional studies should be looked at for potential altered expression in malignant cells. UBXD7 facilitates the degradation of HIF1α, an important promoter of angiogenesis, tumor growth, and metastases. 35 Also, HIF1α overexpression is a frequent occurrence in many different solid tumor types (reviewed in Poon et al. 102 ). Thus, it will be interesting to evaluate if UBXD7 expression is down-regulated in cancer and if this is associated with increased HIF1α levels. Similarly, UBXD8/ETEA has recently been documented to promote NF1 degradation. 60 The NF1 Ras GTPase is mutated in neurofibromatosis type 1, a common genetic disorder that predisposes patients to the development of both benign and malignant tumors of the central nervous system. 103 Its activity (resulting in the dampening of Raf/MEK/ERK- and PI3K/AKT-signaling pathways) is down-regulated by multiple mechanisms in sporadic brain cancer as well, including by increased proteasomal-mediated degradation in gliomas. 104 Although we have very little understanding of the ubiquitin system components that modulate NF1 turnover, it will be interesting to see if components such as ubiquitin ligases for NF1 or UBXD8/ETEA are overexpressed in tumors that have accelerated NF1 degradation.
p97 and Adaptors as Anticancer Targets?
Considering a possible role for p97 in promoting cancer cell survival, its expression in cancer cells, and the fact it is an enzyme and can be inhibited by small chemical inhibitors, 105 it may represent an attractive candidate for therapeutic intervention. Considering that p97 function is required for the viability of normal and cancer cells, such general inhibitors of p97 are likely to have toxic side effects to normal cells. Similar to proteasome inhibitors, however, there could be a useful therapeutic window that will have clinical utility in certain cancers. A better option perhaps may be to selectively target p97 activities pertaining to the aggresome-autophagy pathway. Tumor cells experience a plethora of stresses that lead to increased protein misfolding, including hypoxia, nutrient deprivation, oxidative stress, and increased mutation rates and, thus similar to inhibitors of heat shock proteins, 106 may display a marked increased sensitivity to compounds that result in the accumulation of toxic protein aggregates. It will be challenging to find compounds that selectively (i.e., by inhibiting the activities of specific adaptors or the binding of these adaptors to p97) block this p97-dependent pathway, but if such compounds can be identified, it will be of interest to assess their antitumor activities and their toxicity to normal cells.
Summary
When looking at the data as a whole, there is currently very little evidence that loss or gain of p97-directed activities plays a direct role in the cellular transformation or the development of cancer. Also, there are currently no mouse modeling experiments demonstrating that loss or overexpression of p97 or its adaptors promotes malignancy in vivo. The best evidence of p97 adaptor involvement comes from studies of FAF1, although we still do not know if its NF-κB’s suppressing function is p97 dependent. As discussed, there is currently much circumstantial evidence linking p97-containing complexes to proliferation control and cancer, but more direct proof is needed. It will not be surprising, however, to find that similar to other components of the ubiquitin system, p97 activities will play a direct role in controlling tumor cell proliferation and are altered in cancer cells. There is also a strong possibility that p97-containing complexes play an important role in maintaining cancer viability through the clearing of potentially toxic misfolded and aggregate-prone material through the aggresome-autophagy degradation pathway. If so, targeting p97 may have therapeutic value. Much investigation lies ahead in terms of unraveling p97 functions, including their its roles in cancer biology and therapy.
Footnotes
D.S.H. is supported by National Institutes of Health (NIH) grant GM070769.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.