
Abstract
The enigmatic MYC oncogene, which participates broadly in cancers, revealed itself recently as the maestro of an unfolding symphony of cell growth, proliferation, death, and metabolism. The study of MYC is arguably most challenging to its students but at the same time exhilarating when MYC reveals its deeply held secrets. It is the excitement of our richer understanding of MYC that is captured in each review of this special issue of Genes & Cancer. Collectively, our deeper understanding of MYC reveals that it is a symphony conductor, controlling a large orchestra of target genes. Although MYC controls many orchestra sections, which are necessary but not sufficient for Myc function, ribosome biogenesis stands out to reveal Myc’s primordial function particularly in fruit flies. Because ribosome biogenesis and the associated translational machinery are bioenergetically demanding, Myc’s other target genes involved in energy metabolism must be coupled with energy demand to ensure that cells can replicate their genome and produce daughter cells. Normal cells have feedback loops that diminish MYC expression when nutrients are scarce. On the other hand, when deregulated Myc transforms cells, their constitutive bioenergetic demand can trigger cell death when energy is unavailable. This special issue captures the unfolding symphony of MYC-mediated tumorigenesis through reviews that span from a timeline of MYC research, fundamental understanding of how the MYC gene itself is regulated, the study of Myc in model organisms, Myc function, and target genes to translational research in search of new therapeutic modalities for the treatment of cancer.
The MYC oncogene contributes profoundly to human cancer morbidity and mortality, and hence the understanding of this gene is essential for the prevention and combat of these devastating diseases. MYC is a prototypical oncogene that has a rich and circuitous history (Wasylishen & Penn, 1 this issue) with surprises lurking around every corner along the journey, leading to our current understanding of this enigmatic gene and its product c-Myc (hereafter termed Myc). 2 In our collective quest to understand the MYC gene, which plays a broad role in cell growth and proliferation in normal, cancer, and stem cells, we need to consider how a cell is made and how MYC influences this process. A cell comprises 55% protein, 25% RNA, 10% lipid, and about 3% DNA, with half of the RNA and protein consisting of ribosomal RNA and proteins. In fact, 50% of ongoing RNA synthesis involves the production of rRNAs. Hence, the making of a cell is largely determined by its ability to undergo ribosome biogenesis, which imposes a significant bioenergetic demand on the cell. 3,4 As the cell grows in the G1 phase of the cell cycle, energy and anabolic building blocks must be concurrently imported to meet the metabolic demand in preparation for crossing the start point and entry into S phase. Upon completion of DNA synthesis that is coupled with surveillance for replication fidelity, the cell further prepares to divide the replicated genome linked to many safeguards in mitosis before splitting into 2 daughter cells. Glucose is the major energy source for cells, and the determination of lactate and carbon dioxide production from glucose in controlled batch cultures has led to an estimate that 2 × 1012 ATPs are required to make a mouse LS cell. 5 As such, a central regulator of cell growth and proliferation such as MYC must have the ability to stimulate energy uptake, biosynthetic events, and cell cycle progression in a temporally coordinated manner within a vast network of other regulatory molecules (Figures 1 and 2).
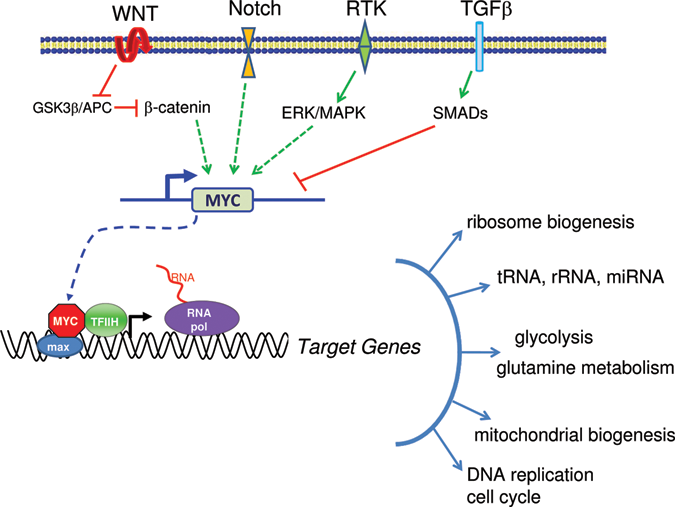
The MYC gene is depicted downstream of different signal transduction pathways, such as WNT, Notch, receptor tyrosine kinases (RTK), and TGFβ. The MYC gene produces the Myc transcription factor, which dimerizes with Max, recruits TFIIH, and stimulates RNA polymerase activity to activate target genes. Myc affects diverse types of target genes regulated by RNA polymerases I, II, and III as illustrated.
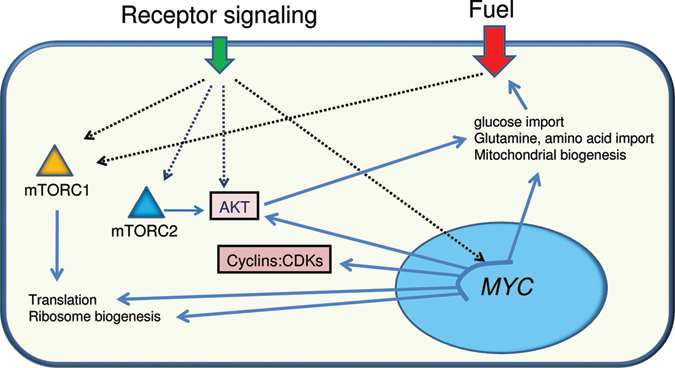
Schema of receptor signaling through cytoplasmic integrators such as mTORC1, mTORC2, and AKT or other relays that stimulate MYC expression. The cytoplasmic integrators through posttranscriptional modification of substrate proteins or Myc via activation of target genes can stimulate a cellular response that increases the uptake of energy and anabolic building blocks to support translation for cell growth and proliferation.
Now that the Myc transcription factor is established as a bona fide conductor of the symphony of metabolism, cell growth, and proliferation (Figure 1), it is not surprising in retrospect that v-myc was among the first oncogenes to be identified because of its pleiotropic effects on many different cellular functions and its central role in tumorigenesis. 6,7 The proto-oncogene MYC is highly regulated under the extreme scrutiny of a large variety of signal transduction pathways culminating in a cast of transcription factors that orchestrate MYC expression through canonical B-DNA binding sites or through more elaborate DNA structures such as the G-quadruplex in regulatory regions of the MYC gene 8 (Levens, 9 this issue). For example, signal transduction through the WNT or Notch pathway results, respectively, in β-catenin or Notch intracellular domain–mediated transcriptional activation of MYC expression (Figure 1). TGFβ, on the other hand, can attenuate MYC expression through Smad transcription factors (Figure 1). Normal MYC is hence part of a highly adaptive and flexible network of many regulatory molecules that are tweaked by cues external to the cell, such that when growth signals abate, receptors and cytoplasmic integrators respond, MYC expression diminishes, and cells become dormant (Figures 2 and 3).
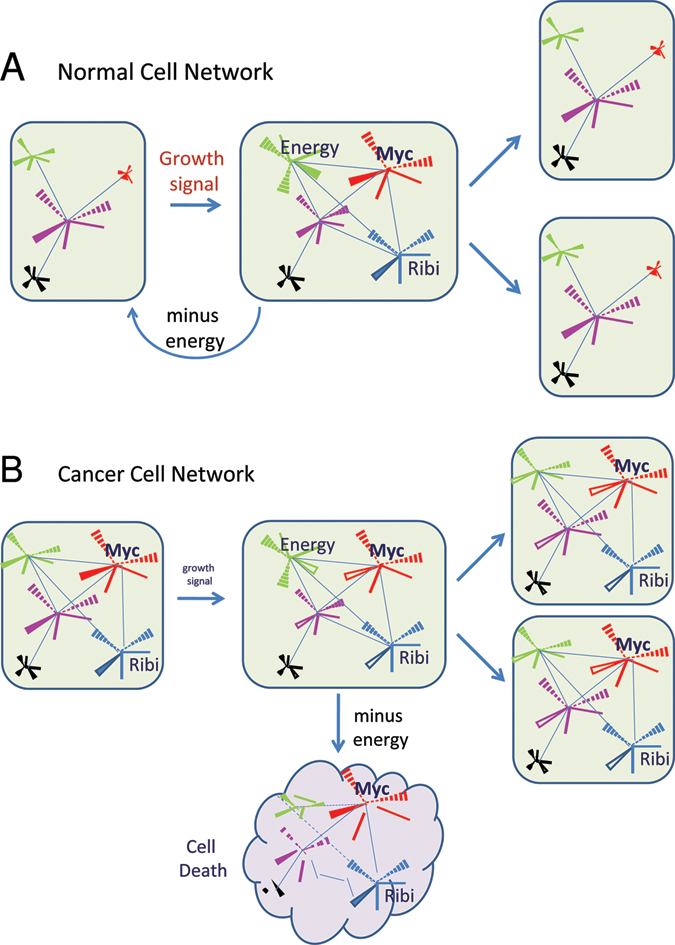
Conceptual normal and cancer cell networks are depicted as starburst nodes linked by edges (lines). (
In the flexible but regulated normal cellular network, the orchestration of regulatory molecules relies on feed-back and feed-forward loops that control their levels through synthesis, posttranslational modification, or degradation in a temporally coordinated manner. 10,11 When normal cells are replenished in damaged tissues by tissue stem cells or in tissues that have a naturally high turnover rate, the extracellular matrix and growth factors engage a variety of receptors to trigger a response leading to increased energy uptake and enhanced biosynthetic events for cell growth in preparation for entry into S phase (Figures 2 and 3). 12 In contrast, when natural mutations result in the activation of oncogenes such as MYC, high levels of Myc trigger checkpoints that eliminate the deranged cell through cell death. However, when accompanying mutations subdue the programmed suicidal tendency so that cells are able to survive even with high MYC levels, cancer cells emerge. 13 In this setting, deregulated MYC reprograms the highly flexible normal network to one that is rigidly linked to heightened MYC activity, which is not subject to the editing function of external cues (Figure 3). In particular, it is envisioned that a sustained increase in MYC activity results in a distorted network of regulatory molecules whose levels and regulation are reprogrammed, such that removal of MYC activity from this altered network could cause an uncoordinated decay of the regulatory nodes, resulting in an imbalanced network that culminates in chaos and cell death, otherwise known as oncogene addiction 14 (Felsher, 15 this issue). What then is the function of Myc, and how does a flexible network with a tunable MYC gene in normal cells differ from one in which MYC is locked at a high volume in cancer cells?
The MYC gene produces a helix-loop-helix leucine zipper transcription factor, Myc, that dimerizes with Max to bind DNA and regulate transcription through recruiting cofactors and initiating transcription or alleviating transcriptional pause. 7,16 Myc also has nontranscriptional roles including its involvement in mRNA capping (Cowling & Cole, 17 this issue) and DNA replication. 18 Further, the Max network of proteins, which can dimerize with Mad proteins to antagonize some of Myc’s function, 19 has related family members such as Mlx, which dimerizes with Mondo proteins, one of which is involved in the regulation of energy metabolism 20 (Sloan & Ayer, 21 this issue). Myc also mediates transcriptional repression through its direct interaction with Miz-1 7 (Herkert & Eilers, 22 this issue), activation of microRNAs 23 (Bui & Mendell, 24 this issue), or yet-undefined mechanisms that require tethering of Myc to noncanonical Myc binding sites. As for its transcriptional role, the target genes of Myc have been a key focus for the field that began with low-throughput subtraction cloning approaches to the current high-throughput use of microarrays and deep sequencing of chromatin immunoprecipitated DNA 25 (McMahon, 26 this issue). Intriguingly, although Myc has been found to bind to thousands of binding sites corresponding to thousands of genes, only a minority of the bound genes respond to Myc, as seen in several experimental systems. 27,28 In this regard, the theme that multiple transcription factors are required to fire off transcription is beginning to emerge with the ability to use high-throughput ChIP identification of binding sites. Even with these powerful technologies, the universe of Myc target genes in a variety of systems is sizeable and bewildering with genes involved in diverse cellular functions.
The network of Myc target genes becomes even more complex with the documentation that Myc also regulates noncoding RNAs. Myc regulates microRNAs, which are 22- to 23-nucleotide long RNAs that modulate mRNA half-lives or translation and can affect up to hundreds of mRNAs 23 (Bui & Mendell, 24 this issue). Moreover, not only does Myc regulate RNA polymerase II genes but it also regulates rRNAs through RNA polymerase I and small RNAs through RNA polymerase III. 29 Thus, Myc uses a variety of tactics to regulate multiple components of the translational machinery, such as tRNAs, aminoacyl-tRNA synthetases, and eIF4E as well as mRNA capping via Myc’s nontranscriptional activity, to ensure that ribosome biogenesis is substantially augmented. 4,30
With an extremely large orchestra of Myc target genes, which of these targets plays the most important part of the MYC symphony? It is doubtful that an accomplished symphony conductor would highlight a certain orchestra section over others, but it is well known that there are 1st and 2nd strings, with the leader of the 1st violin serving as the concertmaster, subordinate only to the conductor. In this regard, each section of the orchestra (brass, percussion, strings, and woodwinds) is necessary but not sufficient for the conductor to execute a symphony. Further, the redundancies in certain sections, such as strings, can accommodate the loss of a single violinist. As such, in the MYC symphony, no individual player is sufficient to replace MYC, 31,32 but there are many key target genes that are necessary for Myc’s role in activating cell growth and proliferation. 33 There are also Myc target genes (such as JTV-1) that are neither sufficient nor necessary, 34 due to redundancies in certain sections of Myc targets. A glimpse into the potential key function of Myc could be gleaned from phylogenetic analysis of conserved Myc function. In this regard, the discovery of Drosophila dMyc opened up a genetic vista into the primordial function of Myc that would otherwise be too complex to swiftly emerge from experiments with mammalian systems 35,36 (Bellosta & Gallant, 37 this issue). Diminutive fruit flies with small body size and shortened bristles are the result of a hypomorphic allele of dMyc that phenocopies loss of function in genes involved in ribosome biogenesis. 38-40 Hence, it is likely that the primordial function of MYC is to stimulate ribosome biogenesis, which could be considered the strings and concertmaster of the MYC symphony.
As microarray data sets emerge from the study of Myc and a related family member, N-Myc, the link between Myc activation and the expression of genes involved in ribosome biogenesis in mammalian cells becomes better established. 3,4,30 The role of ribosomes in tumorigenesis was documented by the delay in Myc-mediated lymphomagenesis and decreased mortality when only 1 copy of RpL24 is available. 41 Because the synthesis of ribosomes is a bioenergetic liability when the demand for energy exceeds availability, feedback loops are built into the network such that glucose deprivation can attenuate rRNA gene expression epigenetically, thereby down-regulating the pace of ribosome production. 42,43 As such, when MYC is deregulated and genes involved in ribosome biogenesis are expressed constitutively in disregard to the availability of nutrients, this rigid network causes a cell with deregulated MYC to be vulnerable to nutrient deprivation (Figure 3). Analogously, a revved-up engine requires effective fuel injection; otherwise, the engine would burn out. Indeed, deregulated MYC expression sensitizes cells to both glucose and glutamine deprivation, which trigger apoptosis. 44-46 So in addition to checkpoints and feedback loops, such as activation of TGFβ signaling, that are activated when Myc is highly overexpressed (Felsher, this issue 15 ), the deregulated expression of MYC could also cause a metabolic demand from the stimulation of ribosome synthesis and other biosynthetic processes, which, if not sustained, will trigger cell death.
The requirement for increased metabolic demand via Myc’s induction of ribosome biogenesis is met by the ability of Myc to induce the expression of genes involved in both glucose and glutamine metabolism 47,48 (Sloan & Ayer, 21 this issue). A key difference between normal and Myc-driven cancer cells is that nutrient withdrawal from normal cells would trigger a network response that causes MYC itself to be silenced, permitting the cell to retreat into a resting state. In contrast, when MYC is deregulated, nutrient withdrawal cannot attenuate MYC levels, which constitutively signal the cell to build and build for growth sake in a manner disconnected from the cellular energetic fiscal reality, much like the overbloated housing market, which crashed with the realization that the financial fuel was just an illusion. Deregulated MYC hence causes Myc-transformed cells to be addicted to nutrients such as glucose and glutamine. 48 It is surmised from the concepts derived here from the study of cells in culture that oncogene addiction, particularly addiction of tumors to Myc in genetically engineered mouse models (Felsher, this issue 15 ), could well be due to a more rigid network that drives cells to grow and become more dependent on nutrients. In this regard, removal of Myc is accompanied by an asynchronous tuning down of the cancer cell’s regulatory network, culminating in an imbalance in energy availability versus energy demand and resulting in cell death. Analogously, when the orchestra director prematurely leaves the hall at the peak of a crescendo, the beat does not go on, but rather the orchestra tumbles in chaos and the symphony cacophonously disintegrates.
The understanding of Myc function from the study of cells and model organisms (Bellosta & Gallant 37 and Felsher, 15 this issue) has led to better insights into the roles of Myc in human cancers such as leukemias 49 (Delgado & León, 50 this issue), breast cancer 51,52 (Xu et al., 53 this issue), and prostate cancer 54 (De Marzo et al., 55 this issue). In all of these different settings, a common thread that putatively connects these cancers is the occurrence of hypertrophied nucleoli, the site for ribosome biogenesis, in the more aggressive cancers of each type. 56 With MYC driving a significant portion of human cancers and the realization that many of these cancers might be addicted to MYC, could Myc or its target genes provide novel therapeutic opportunities? In this regard, targeting MYC expression itself through pharmacological manipulation of the G-quadruplex 57 (Hurley & Brooks, 58 this issue), drug-mediated disruption of Myc-Max protein-protein interaction 59-61 (Prochownik & Vogt, 62 this issue), or manipulation of microRNA Myc target genes themselves hold promise for new classes of anticancer drugs 63 (Frenzel et al., 64 this issue).
Although novel approaches have permitted a glimpse of the conductor and wizard behind the curtain, the enigmatic MYC oncogene is likely still to hold back some deeply held secrets from our quest to understand its contribution to normal, cancer, and stem cell growth and proliferation. For example, although our understanding of how Myc stimulates cell growth and proliferation is coming into sharper focus, our knowledge of how Myc collaborates with other transcription factors 65 and how Myc suppresses terminal differentiation remains rudimentary. 66,67 Further, whether chromosome 8q24–associated cancers will be susceptible to therapies targeting MYC remains to be delineated (Grisanzio & Freedman,68 this issue). Our substantial understanding of the MYC oncogene, some 30 years after its discovery, has established a foundation to begin to translate basic observations about this gene for cancer therapy in the clinic. 64 Perhaps there will be a branch in the road ahead, where taking the one not taken might reveal some additional secrets held by the enigmatic MYC oncogene.
Footnotes
Acknowledgements
I limited many of the references to review articles and hence unintentionally omitted important primary articles. I thank Debbie Johnson and Vanessa Dang for comments.
Our original work is supported by NCI, NIH, Leukemia and Lymphoma Foundation, and the Stand-Up-to-Cancer AACR initiative.
C.V.D. is a consultant for Agios Pharmaceuticals, Inc.