
Abstract
The iconic history of the Myc oncoprotein encompasses 3 decades of intense scientific discovery. There is no question that Myc has been a pioneer, advancing insight into the molecular basis of cancer as well as functioning as a critical control center for several diverse biological processes and regulatory mechanisms. This narrative chronicles the journey and milestones that have defined the understanding of Myc, and it provides an opportunity to consider future directions in this challenging yet rewarding field.
As one of the first oncogenes identified, Myc has enjoyed an illustrious history (see Figure 1). In reflection of 30 years of research and in excess of 20,000 published articles, 2 major themes emerge. First, the fine-tuned regulation and numerous activities of Myc in normal cells are unparalleled and a beauty to behold. Second, when this regulation is lost, Myc rears its beastly head and takes the role of one of the most prominent and intriguing oncoproteins. This review focuses on the history and early work that has defined Myc. It also provides perspective for the articles that follow, which will comprehensively discuss Myc in its current context.
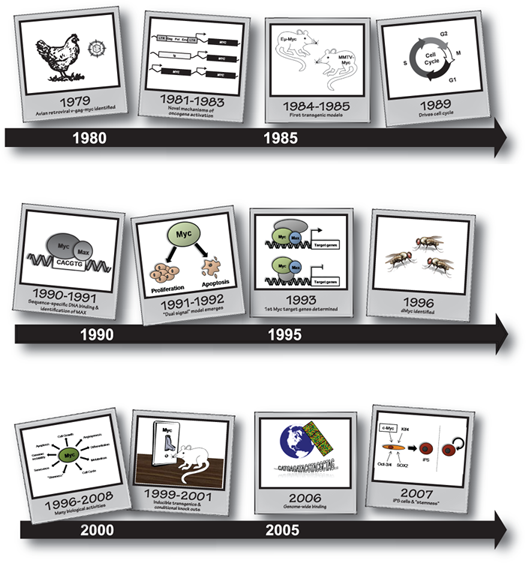
Snapshots from Myc’s history.
MYC Is Established as a Prominent Oncogene
Identification of MYC
In the late 1970s, an avian acute leukemia virus (MC29) was shown to promote a spectrum of malignancies, including myelocytomas, sarcomas, and importantly, carcinomas. This ability to induce carcinomas was of particular interest because this represented the most common form of human malignancy. The transforming sequence of MC29 was identified as v-myc and named myelocytomatosis for a resultant leukemia. v-myc was found to present in the cells as a 110-kDa v-gag-myc fusion. Consistent with the notion that oncogenes could be stolen by retroviruses, the cellular homologue was identified soon after in normal cells from many species. The discovery of MYC further reinforced the startling realization that oncogenic transformation could be caused by the activation of a cellular gene. 1–4
Mechanisms of Deregulation in Cancer
Building on the awareness that, unlike other proto-oncogenes, MYC activation was not a consequence of mutations in the coding sequence, research focused on identifying and understanding other modes of oncogenic activation. This led to the discovery of 3 novel mechanisms through which Myc and, in turn, other oncogenes could be deregulated and promote transformation: insertional mutagenesis, chromosomal translocation, and gene amplification. Combined, these findings led the way for the discovery and understanding of oncogenes and provided new paradigms for the genetic basis of cancers.
In addition to the acutely transforming retrovirus mentioned previously, a second class of retroviruses had been shown to induce leukemias and lymphomas. These viruses, often referred to as non–acutely transforming retroviruses, induced tumors with a much longer latency and were unable to transform cells in culture. Although these initial observations were puzzling, the mechanisms through which these viruses promoted tumorigenesis were soon realized when retroviruses were shown to activate the expression of oncogenes through proviral insertion. 5–8 Specifically, viruses such as the acute leukosis virus (ALV) integrated into the host genome at or near proto-oncogenes, resulting in a high level of expression driven by the viral promoter. Myc was the first oncogene found to be activated by this mechanism, with 80% of B-cell lymphomas induced by ALV owing to activated MYC. These groundbreaking studies laid the foundation for the discovery of several other oncogenes. 9
Nonrandom chromosomal translocations had been observed in a number of malignancies, including Burkitt lymphoma and chronic myeloid leukemia. It was tempting to speculate that these translocations resulted in aberrant expression of the same proto-oncogenes identified in the acutely transforming retroviruses. The mapping of MYC to the long arm of chromosome 8 gave creed to this hypothesis. 10,11 Specifically, Burkitt lymphomas had been characterized to contain reciprocal translocations between chromosome 8 and chromosome 14, 2, or 22, which harbor immunoglobulin (Ig) heavy and light chain genes. 12 It was then discovered that these cancers were driven by activated expression of MYC resulting from the translocation. The first MYC transgenic mouse, Eµ-Myc, was developed to model Burkitt lymphoma, with activated MYC expression driving a clonal B-cell lymphoma. 13 Mouse plasmacytomas were similarly found to be a consequence of MYC translocation with the Ig heavy chain locus. 14,15
It was well established that cancer cells contained a number of chromosomal abnormalities, including the presence of double minute chromosomes and homogeneously staining regions. The contributions of these aberrations to cellular transformation were largely appreciated through the study of MYC. Human colon cancer (COLO-320) and acute promyelocytic leukemia (HL-60) cell lines were shown to express multiple copies of MYC. 16–18 Importantly, gene amplifications were observed in primary patient material, including uncultured samples from the patient whose tumor was the source for HL-60 cells. 17 Overall, these findings further supported the understanding that deregulated expression of a proto-oncogene could promote neoplastic transformation.
A Family Affair
Through these studies, it became clear that MYC was a member of a larger family of oncogenes. When v-myc was used as a probe in hybridization experiments, additional restriction fragments were consistently and reproducibly identified, which suggested that the human genome contained a series of sequences that were similar or related to the MYC oncogene. Amplification of a new MYC family member was soon identified in a panel of neuroblastoma samples and cell lines. 19,20 This new oncogene was named MYCN, for the neuroblastomas in which it was identified. Importantly, MYCN was quickly associated with aggressive disease and poor outcomes in this pediatric cancer, and amplifications were assigned great prognostic value. 21,22 MYCN amplification in neuroblastoma provided one of the earliest examples of how basic research involving oncogenes could be translated into a clinically relevant prognostic marker, a principle that remains at the forefront of cancer research today.
Within an additional couple of years, a third family member was identified through gene amplifications in small-cell lung cancer and was fittingly named MYCL1. 23 Further research identified amplifications of these 3 transforming members of the Myc family in a number of other tumor types. 24 These amplification studies expanded the role for the Myc family in the pathogenesis of a large number and wide variety of human malignancies, beyond leukemias and lymphomas.
With a Little Help from MYC’s Friends
MYC was also fundamental to the understanding of oncogene cooperation and how cellular oncogenes could promote tumorigenesis. These foundations were built through studies in primary rat embryo fibroblasts. 25 Ectopic expression of activated RAS (EJ-RAS) alone was insufficient to promote transformation; however, cotransfection of EJ-RAS and v-Myc or c-Myc promoted foci formation in vitro, which provided the first evidence that cooperation between cellular oncogenes was essential for cellular transformation. This cooperation model was supported by observations from MYC transgenic models. The tumors that arose in Eµ-Myc mice were clonal in origin and thereby suggested a need to acquire a secondary mutation for tumor development. 13 When Eµ-Myc mice were crossed with Eµ-Bcl2 mice, driving constitutive overexpression of the antiapoptotic BCL2 oncogene, tumors formed more rapidly than the single transgenic. 26,27 The model of oncogene cooperation and the need for multiple collaborating alterations to promote tumorigenesis have become fundamental to our current understanding of the neoplastic process. 28
Myc Regulation in Nontransformed Cells
It was clear that there were multiple modes through which deregulated Myc expression could be achieved, which shifted the focus to understanding the normal regulation of Myc. Serum stimulation of quiescent cells in culture resulted in a rapid upregulation of Myc mRNA. 29 The induction peaked at 2 hours poststimulation, was cycloheximide independent, and established MYC as an intermediate–early response gene. The converse was also true that antiproliferative signals resulted in rapid downregulation of Myc. 30–33 Additionally, Myc mRNA and protein demonstrated short half-lives. 34,35 As such, an appreciation was developed for the tight regulation of Myc expression in non-transformed cells, which was exceptionally responsive to extracellular cues. Because Myc is one of the most highly regulated proteins in the cell, many important regulatory mechanisms have been discovered through studies of Myc.
Transcriptional Control and mRNA Turnover
From early on, considerable research efforts were directed at understanding the regulation of MYC at the transcriptional level. The gene was shown to have rather unusual topology, with a large noncoding first exon and 2 additional coding exons (reviewed in References 36 and 37). Within the 5′ region of exon 1, transcription can be initiated from 2 promoters (P1 and P2) separated by 150 base pairs, with the majority of transcripts arising from P2. The complex array of regulatory mechanisms and the multitude of signaling cascades that converge on MYC have made this a challenging area of research. MYC, however, has maintained its role as a pioneer and was the first eukaryotic gene found to be regulated by transcriptional elongation control. 38–41 There is clearly much remaining to be discovered, and recent studies suggest that we are now in a strong position to better delineate some of these mechanisms. For example, through their interactions with the far upstream element (FUSE; 1.7 kb upstream of P2), FUSE-binding protein (FBP) and FBP-interacting repressor have recently been identified as important players in regulating transcription of MYC. 42,43 Additionally, genomewide association studies have linked single-nucleotide polymorphisms on chromosome 8q24, upstream of MYC, to an increased risk of some cancers. 44–49 Further studies will continue to advance our understanding of the transcriptional regulation of MYC.
Protein Expression and Regulation
Myc has 2 primary protein products that arise from an AUG codon at the 5′ end of exon II and a CUG initiation codon at the 3′ end of exon I that can be visualized as 64-kDa and 67-kDa proteins by immunoblot analysis. 37,50,51 An additional short isoform, MycS, that lacks the first 100 amino acids has been reported 52 ; however, the functional and/or regulatory role of this molecule is not fully understood. Tryptic phosphopeptide mapping has revealed a number of serine and threonine sites within Myc that are phosphorylated in vivo. 53–55 Of these, serine-62 (S62) and threonine-58 (T58) have dominated our understanding of Myc posttranslational modifications. 56,57 Much of the interest in these residues has been driven by the identification of point mutations in the homologous threonine of v-Myc (T61) and in the detection of point mutations at S62 and T58 and surrounding residues in some cases of Burkitt and AIDS-associated lymphomas. 58–61 Collectively, research has shown that S62 is phosphorylated in response to proliferative signals, which are thought to stabilize and promote c-Myc activation. Phosphorylated S62 then allows GSK3 to bind and phosphorylate T58, which subsequently promotes binding of FBXW7 and recruits the SCF complex, leading to the ubiquitylation and proteosomal degradation of c-Myc. Interestingly, many regulators of Myc are themselves oncogenes and tumor suppressor genes, and targeting these signaling pathways provides an indirect approach to inhibit Myc activity. In addition, c-Myc has been shown to be O-glycosylated and acetylated, although our understanding of the biological role of Myc posttranslational modifications remains in its infancy. This is remarkable for a protein like Myc, which has been at the forefront of cancer research for decades, and it is clear that this will be an important area of future work.
The Biological Activities of Myc
Cell Cycle and Differentiation
That the deregulation of proto-oncogenes was fundamental in tumorigenesis made it natural to speculate that they functioned in normal cell proliferation. There were several lines of evidence to suggest that Myc might play a role in cell cycle progression. As mentioned previously, MYC was established as an intermediate–early response gene when serum stimulation of quiescent cells rapidly induced Myc mRNA. 29 Additionally, ectopic expression of Myc promoted growth factor–independent proliferation of mouse fibroblasts in culture. 62 More definitive evidence came several years later through the use of conditional Myc–estrogen receptor (Myc-ER) chimeras, in which Myc was fused to the hormone-binding domain of the ER. 63 Activation of Myc-ER by estradiol promoted cell cycle entry of quiescent cells in tissue culture. 64 Therefore, it was clear that Myc functioned in promoting cell cycle progression.
Not unrelated to the ability to promote cell cycle progression, Myc expression was shown to block differentiation. It was demonstrated by multiple groups and through a number of models that Myc downregulation was essential for cells to exit the cell cycle and undergo differentiation. For example, ectopic expression of Myc was shown to block the differentiation of 3T3-L1 preadipocyte, 65 MEL murine erythroleukemia, 66–68 and F9 teratocarcinoma 69 cell lines. These abilities to promote cell proliferation and block differentiation have natural associations with tumorigenesis and are features of aggressive disease.
Apoptosis
With well-established contributions to tumorigenesis and convincing evidence to suggest that Myc played a role in cell cycle and proliferation, the field was met with much surprise in the early 1990s when ectopic expression of Myc was shown to sensitize cells to undergo apoptosis. 70–72 It was initially perplexing—why and how could a single molecule drive such disparate functions? As was the case previously, this groundbreaking observation for Myc soon held true for other oncogenes, such as E2F1 and E1A. 73 With this, a dual-signal model emerged. Myc was able to encourage both proliferation and apoptosis. Apoptosis, however, could be averted by survival signals, including those from insulin- and platelet-derived growth factors. 72,74 Myc’s ability to promote apoptosis provided a built-in safety mechanism to protect against inappropriate proliferation as a consequence of deregulated Myc. This finding shed considerable light on the model of oncogene cooperation and how an antiapoptotic protein such as Bcl2 could cooperate with Myc to promote oncogenesis. 27,75,76 Specifically, it suggested that abrogating Myc-induced apoptosis was a critical component of cellular transformation.
Within a few years, the field had a better appreciation for some of the mechanisms through which Myc could drive apoptosis. Deregulation of Myc was shown to activate the tumor suppressor p53 through the upregulation of ARF. 77–79 A loss of either of these tumor suppressors accelerated tumorigenesis in mouse models of oncogenic Myc. 80–84 Other regulators of the ARF-MDM2-p53 axis have been shown to cooperate with Myc to promote cellular transformation. 85–87 Furthermore, Myc is able to promote apoptosis through p53-independent mechanisms by influencing the balance between proapoptotic and antiapoptotic proteins in the cell. 73,88–94 Combined, there is much interest in improving our understanding of the mechanisms through which Myc promotes apoptosis in the cell and how this activity is abrogated in cancer.
Molecular Mechanisms
Although it was vital to determine Myc’s biological activities in the cell, it was equally important to understand its mechanisms of action. Early work studying v-gag-myc using antibodies against gag demonstrated that, unlike many other oncogenes at that time, Myc was localized to the nucleus and able to bind double-stranded DNA. 95,96 Soon after, this observation was confirmed for the native protein. 97 From this, 2 models emerged. First, Myc was hypothesized to play a role in DNA replication. Second, Myc was believed to regulate gene transcription, either directly or indirectly. It was the latter model that soon took center stage.
Transcriptional Activation by Myc
Unlike in the first decade of research—in which Myc was at the leading edge, forging new territory and serving as a model for the understanding of other oncoproteins—the field relied on previous work on other proteins with similar domains when it came to understanding the mechanisms of Myc activity. The C-terminus of Myc was shown to contain both a helix-loop-helix 98 domain and a leucine zipper domain. 99 At the time, it was unusual for a protein to contain both these domains, but it encouraged the field to focus on potential protein and DNA interactions. Importantly, through deletion and insertion mutants, these regions were found to be essential for all functional readouts of Myc activity: REF focus formation and Rat1A colony formation, negative autoregulation, and the inhibition of differentiation. 100
The parallel discoveries of a specific DNA-binding sequence for Myc provided a much-needed boost to researchers in the field. 101,102 The first report showed that homodimers of Myc C-terminal domains could bind to CACGTG sequences in vitro. This work and the low affinity with which these dimers formed suggested that Myc must have a partner. The search was on, and soon after Max (Myc-associated factor X) was identified. 103 Max was shown to be a constant and obligate partner for Myc, with consistent and abundant expression in proliferating and quiescent cells, which was not altered in response to extracellular stimuli. It is important to note that Max also forms heterodimers with members of the Mxd family, thereby providing an additional mechanism to regulate Myc activity in the cell. 104,105
The first direct evidence of Myc as a transcriptional activator came from fusion of the Myc N-terminal domain to the yeast Gal4 DNA-binding domain. 106 These fusions were able to activate expression from a reporter construct. The ability of Myc to function as a transcriptional regulator was advanced with the discoveries of other interacting proteins, which were in turn components of chromatin-remodeling complexes. For example, the N-terminal domain of Myc was shown to interact with TRRAP and subsequently recruit histone acetyl transferase activity. 107,108
Transcriptional Repression by Myc
Some of the initial evidence showing that Myc was able to function as a transcriptional repressor emerged from studies of Myc expression that suggested negative autoregulation. Specifically, several reports demonstrated that Myc was not expressed from the nontranslocated allele in Burkitt lymphoma. 109,110 It was further shown that expression of v-myc was able to downregulate expression of endogenous Myc, and the same was true for ectopic Myc expression. 111,112 Structure–function studies then revealed that the regions essential for transformation were the same as those required for this negative autoregulation. 113,114 Despite these early observations, the mechanisms of transcriptional repression by Myc remained elusive for some time. In due course, repression was shown to be independent of binding to E-boxes. 115 Additionally, bona fide repressed targets and key interactors were identified that developed our current understanding of transcriptional repression by Myc. For example, MYC-interacting zinc finger 1 (Miz1) was identified as an interacting protein and important mediator of Myc function. 116,117 As such, the current model is that Myc–Max heterodimers interact with transcriptional activators, such as nuclear factor Y and Miz1, which are directly bound to DNA through enhancer or initiator elements. 118,119 This binding displaces coactivators and recruits corepressors, thereby preventing transcriptional activation. Even though the understanding of Myc as a transcriptional repressor lagged behind its role in activating gene transcription, it is clear that this activity is important for Myc function.
Target Genes
Not only was the development of the Myc-ER fusion instrumental in defining Myc’s role in driving the cell cycle, but it also provided a tool for the discovery of the first Myc target genes. 64 The activation of Myc in the presence of cycloheximide to block protein synthesis identified α-prothymosin as a transcriptional target of Myc. This same approach was used to identity a number of other target genes, including ornithine decarboxylase 1. 120,121 Another valuable approach used to identify Myc targets employed mitogen stimulation of quiescent Myc-null rat fibroblast cells to determine if transcriptional regulation was dependent on Myc. 122 This method subsequently demonstrated CAD and GADD45A as activated and repressed Myc targets, respectively. 123–125 Although gene-by-gene approaches were successful overall, identification of Myc targets was slow and laborious. 126
Genomewide approaches and expression microarrays transformed our ability to identify Myc target genes. This was met with its own challenges, however, in that consistency between studies was quite poor. One of the major explanations was that Myc induced only small changes in the mRNA abundance of targets, which made their identification difficult. 127 This prompted the field to establish criteria for designating Myc target genes and to develop a database to catalog and manage genes based on these criteria (http://myc-cancer-gene.org/).
More recently, new technologies have been used to survey Myc binding to chromatin in cells, including DNA adenine methyltransferase identification and chromatin immunoprecipitation (ChIP). These techniques have revolutionized our ability to evaluate protein–DNA interactions in vivo, and they have further improved our ability to identify direct Myc target genes. Additionally, the specificity afforded by ChIP assays, combined with the high-throughput capabilities of microarrays (ChIP-chip) or high-throughput nucleotide sequencing (ChIP-PET and ChIP-seq), has provided the field with the ability to interrogate genomewide binding of Myc. Data from these studies suggest an expanded role for Myc. Binding 10% to 15% of the genome, Myc has been shown to regulate the expression of genes encoding noncoding RNAs, as well as proteins. 127,128 The identification of the direct target genes has started to shed some light on how Myc is able to promote such an array of cellular processes. For example, Myc promotes the cell cycle by activation of cyclins D1, D2, E1, and A1, as well as the repression of p21, p27, p15, and GADD45. However, it has become clear that Myc does not act solely as a classic transcription factor, regulating a small number of target genes to promote a given biological function. Rather, Myc can globally influence chromatin structure and affect genetic programs. 129 We direct you to several excellent reviews that recently discussed Myc regulation of target genes and chromatin (including articles in this issue by McMahon 130 and Bui & Mendell 131 ). 2,127,129,132,133
New Biological Activities Are Added to Myc’s Arsenal
By the late 1990s, Myc had added several different biological activities to its collection and was shown to regulate cell size, affect genomic stability, and trigger the angiogenic switch. First, Myc demonstrated an ability to promote cell growth both in vitro and in vivo by supplying the cell with several classes of basic building blocks and increasing protein synthesis and cell metabolism. 134,135 Many of the universally regulated Myc target genes supported this activity, including those involved in ribosomal and mitochondrial biogenesis, cellular metabolism, and protein and nucleic acid synthesis. 136,137 Second, cells with deregulated Myc were shown to have a higher rate of specific gene amplifications, and further studies indicated that Myc could promote chromosomal instability. 138–141 Although this activity seems to be context dependent, several mechanisms have been proposed, including alterations in chromosome structure, overcoming the p53 checkpoint, and increased levels of reactive oxygen species. 142,143 The third of these novel activities is the ability of Myc to stimulate neovascularization, which has been evident in a number of in vivo models. 144–147 The downregulation of the angiogenesis suppressor thrombospondin and release of interleukin 1β have been shown to play a role in this process. As such, there is no question that Myc’s ability to influence these hallmarks would contribute to tumorigenesis, and we recommend several recent review articles for thorough discussions of these activities. 2,132,143,148
In recent years, additional biological activities of Myc have been characterized. The renewed interest in the Warburg effect and tumor cell metabolism has highlighted a new role for Myc. In addition to stimulating mitrochondrial biogenesis, oncogenic levels of Myc have been shown to promote glutaminolysis. 136,149-151 This increased glutamine metabolism fuels cell growth and proliferation, which are essential for tumor cells to thrive. There have also been suggestions that tumor cells become addicted to glutamine, which may provide opportunities for therapeutic intervention. 136
Although oncogene-induced senescence had been widely recognized as a mechanism through which normal cells protect themselves against transformation, it was only recently that this function was assigned to Myc. Unlike Ras, overexpression of Myc alone does not induce senescence. Myc can, however, induce senescence in the context of the loss of genes such as WRN or CDK2. 152-156 Future research will inevitably explore if Myc-induced senescence can be harnessed to promote tumor regression.
The ability of Myc to block differentiation perhaps foreshadowed the recently uncovered role of Myc in regulating stemness. Conditional knockout mice have demonstrated an essential role for Myc in the normal developmental control of hematopoetic and neuronal stem cells. 157,158 Myc has recently been identified as 1 of the 4 genes whose overexpression could reprogram normal terminally differentiated fibroblasts into induced pluripotent stem cells. 159,160 Although it was later shown that Myc was dispensable for this process, 161 it did underscore important implications for deregulated Myc in initiating and maintaining tumor stem cells. This speaks to the observation that the stem cell signature of undifferentiated and aggressive tumors carries great similarity with the phenotypes of Myc-activated tumors. With such an impressive array of biological activities, the profound impact of Myc deregulation in cancer is even more apparent.
Prospects for Taming the Beast
Given the well-established and widespread role of Myc in promoting tumorigenesis, the question becomes, can we exploit this potent oncoprotein for the diagnosis and treatment of cancers? Inducible transgenic models have demonstrated that even a brief inactivation of Myc can have a profound and cell type–specific effect on tumor cell fate. 162 Importantly, these models have demonstrated that targeting Myc activity is associated with tumor regression. Despite this, translating our understanding of Myc for clinical application has been met by several hurdles. First, the diverse mechanisms through which the widespread deregulation of Myc occurs in cancers preclude traditional molecular diagnostics. As our understanding of the molecular mechanisms of Myc activities continues to advance, it may become possible for molecular pathologists to score deregulation based on activity. Second, the Myc protein is not a classically “druggable” molecule, because it is localized in the nucleus and does not possess enzymatic activity that can be readily blocked. Approaches to target the regulation of Myc or the effectors of Myc activity may provide alternative means to inhibit this oncoprotein. There have been additional concerns about a therapeutic window, given that Myc is essential in all cells. Recent work has directly addressed this important issue and established Myc as a genuine molecular target in cancer. Briefly, a mutant basic helix-loop-helix leucine zipper domain referred to as Omomyc was developed and shown to bind to and inhibit transcriptional activation by Myc. 163,164 In preclinical studies, Omomyc eradicated tumors in a mouse model of Ras-induced lung adenocarcinoma. Moreover, the systemic inhibition of Myc was well tolerated by normal tissues, and all side effects were rapidly reversible. We direct readers to several review articles for further discussions of Myc as a therapeutic target in cancer, 165,166 including those in this issue by Frenzel, Lovén, & Henriksson 167 as well as by Prochownick & Vogt. 168 In sum, targeting Myc activity in cancer is an exciting avenue of current research and has great potential to impact patient care.
Concluding Thoughts
In reflection of just over 30 years of research, there is no question that Myc has been and will continue to be an influential and significant molecule. Many seminal discoveries have been made through studying Myc, and we hope that this review highlights both the beauty and the beast that is Myc.
Footnotes
Acknowledgements
We would like to thank all of our colleagues who have shared in the fascinating and challenging study of Myc, and we apologize to those whose work we were unable to include because of space constraints. We additionally thank the members of the Penn Lab for helpful discussions and critical review of this article.
This work was supported by grants from the Canadian Cancer Society Research Institute, the Natural Sciences and Engineering Research Council of Canada, and the Canadian Institutes of Health Research (FRN CPG99377) (L.Z.P.), as well as a Canadian Breast Cancer Foundation–Ontario Region Doctoral Fellowship (A.R.W.). Additional support was provided by the Ontario Ministry of Health and Long Term Care. The views expressed do not necessarily reflect those of the ministry.
The authors declare no conflicts of interest with respect to the publication of this article.