
Abstract
The concept of inhibiting tumor neovessels has taken the hurdle from the bench to the bedside and now represents an extra pillar of anticancer treatment. So far, anti-angiogenic therapy prolongs survival in the order of months in some settings while failing to induce a survival benefit in others, in part because of intrinsic refractoriness or evasive escape. This review provides an update on recent mechanisms via which tumor and stromal cells induce resistance and discusses recent evolutions in the (pre)clinical development of novel third-generation anti-angiogenic agents to overcome this problem.
VEGF-Targeted Therapy: Successes
It is well established that the angiogenic switch is a critical step in carcinogenesis; clinical successes of angiogenesis inhibitors validate this paradigm. 1,2 Because of the key role of vascular endothelial growth factor (VEGF), anti-angiogenesis research in the past has been focused largely on this molecule. 3,4 This has resulted in Food and Drug Administration (FDA) approval of first-generation VEGF receptor inhibitors (VEGF(R)Is), such as the monoclonal anti-VEGF antibody bevacizumab (Avastin®, Genentech, South San Francisco, CA). A combination of this VEGF inhibitor with chemotherapy prolongs survival of patients with metastatic colorectal and advanced lung cancer 5,6 and progression-free survival (PFS) of patients with metastatic breast cancer. 7 A recent observational study in patients with metastatic colorectal cancer further shows that continuation of bevacizumab while changing the chemotherapeutic regime beyond disease progression leads to an overall survival gain of 12 months, compared to patients in whom treatment was continued with chemotherapy alone. 8
Monotherapy with second-generation broad-spectrum VEGF receptor tyrosine kinase inhibitors (RTKIs), such as sunitinib (Sutent®, Pfizer, New York City, NY) or sorafenib (Nexavar®, Bayer and Onyx, Emeryville, CA), prolongs PFS of renal cancer patients 9,10 and overall survival of patients with advanced hepatocellular carcinoma 11 or imatinib-resistant gastrointestinal stromal tumors (GISTs). 12 For more information about the mechanisms of these anti-angiogenic drugs and the results of the clinical trials with these agents, we refer the reader to recent reviews. 2,3,13
VEGF-Targeted Therapy: Challenges
Translating the preclinical successes into clinical practice, identifying the optimal clinical indications, and maximizing the efficacy of anti-angiogenic therapy for cancer patients have been more challenging than anticipated. For instance, different from what preclinical studies predicted, bevacizumab improves clinical results only/mostly when combined with chemotherapy, in part because it “normalizes” residual tumor vessels and improves drug delivery. 14,15 In contrast, RTKIs are capable of suppressing tumor growth as monotherapy, 2 likely because they promiscuously inhibit multiple targets. Also, the clinical benefit of VEGF-targeted therapy variably depends on the tumor type, stage, and treatment history. Another example is the recent finding that, unlike promising preclinical results, 16 combination therapy of cytotoxic agents with bevacizumab plus anti–epidermal growth factor receptor (EGFR) shortened PFS of patients with colorectal cancer when compared to bevacizumab and chemotherapy alone, because of unknown reasons. 17,18
Another hurdle is that VEGF(R) inhibitors cause endothelial dysfunction and vessel pruning in healthy tissues, which underlies the adverse effects in a small fraction of cancer patients. 5,9,10,19-23 Although usually manageable and rarely outweighing the clinical benefit, these side effects can become aggravated when bevacizumab and RTKIs are combined 24 and, in certain cases, may necessitate dose reductions, drug holidays, or discontinuation of therapy. 22,25 This safety profile warrants caution against systemic use of VEGF(R) blockers in children, pregnant women, and patients at risk for adverse effects.
But perhaps the greatest challenge today is the fact that a substantial number of cancer patients are intrinsically refractory to anti-angiogenic drugs and do not respond at all or only minimally, 2 such as pancreatic cancer patients. 26 Other cancer patients, who initially respond, escape from anti-angiogenic therapy and experience a survival benefit in the range of months but without permanent cure. 2 Thus, despite transient disease stabilization or tumor regression by VEGF(R)Is, reflected by prolongation of PFS, overall survival is often not (or only negligibly) prolonged in these individuals. For instance, bevacizumab plus paclitaxel therapy of metastatic breast cancer patients or bevacizumab monotherapy of renal cancer patients prolongs PFS but not overall survival. 7,27
Also, the consequences of anti-angiogenic therapy seem to be short-lived, as withdrawal from such treatment induces rapid regrowth of tumor vessels and relapse of tumor growth. Preclinical studies with VEGF(R) inhibitors indeed demonstrate that tumor vessels regrow rapidly after treatment arrest, by tracking alongside empty basement membrane sleeves of ghost vessels. 28,29 Similarly, the beneficial effects of a pan-VEGF receptor tyrosine kinase inhibitor on tumor growth, vessel permeability, and intracranial edema were rapidly lost in glioblastoma patients during brief drug holidays for only a few days. 25
So far, it has been challenging to match optimally the pharmacological profile of an anti-VEGF(R) drug with patient selection or to readjust anti-angiogenic therapy upon disease progression. This is in part attributable to the reality that we insufficiently understand the mechanisms underlying refractoriness and evasive escape, not only in the mouse but certainly in cancer patients, in whom multidisciplinary translational studies are sorely required. 30 Another drawback is the lack of well-validated biomarkers to monitor efficacy and optimal dosing or predict toxicity or resistance to VEGF-targeted therapy. 31 Such biomarkers would offer opportunities to tailor optimal successful anti-angiogenic therapy for individual cancer patients and avoid unnecessary ineffective but costly anti-angiogenic therapy for nonresponders. 32 Table 1 overviews different classes of biomarkers for the efficacy of anti-angiogenic therapies, many of which need to be confirmed independently and validated in phase III clinical trials (see Supplementary Table 1 online for more details). 33
Potential Biomarkers of Benefit of Anti-Angiogenic Therapy
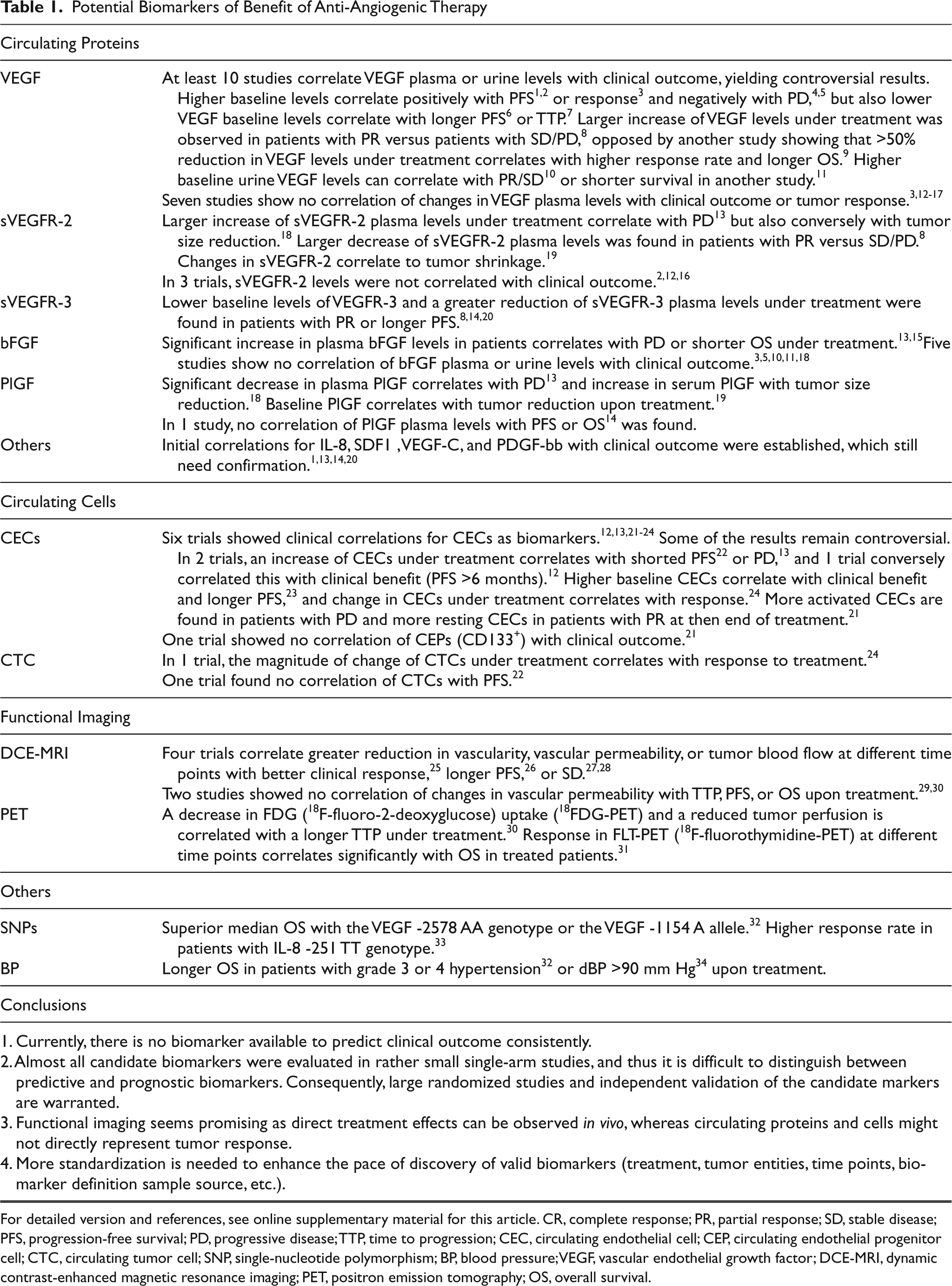
For detailed version and references, see online supplementary material for this article. CR, complete response; PR, partial response; SD, stable disease; PFS, progression-free survival; PD, progressive disease; TTP, time to progression; CEC, circulating endothelial cell; CEP, circulating endothelial progenitor cell; CTC, circulating tumor cell; SNP, single-nucleotide polymorphism; BP, blood pressure; VEGF, vascular endothelial growth factor; DCE-MRI, dynamic contrast-enhanced magnetic resonance imaging; PET, positron emission tomography; OS, overall survival.
An increasing list of emerging preclinical studies revealed insights in the mechanisms of the intrinsic refractoriness and evasive escape (acquired resistance) from anti-VEGF agents. Some of these mechanisms are attributable to the tumor cells themselves (Figures 1 and 2), but malignant cells also seem to highjack their microenvironment to promote escape from VEGF-targeted therapy (Figure 2). Hence, normalization of the microenvironment may offer new avenues to develop third-generation anti-angiogenic drugs (Figure 3). 34 In the next sections, we discuss these anti-angiogenic resistance mechanisms in more detail.
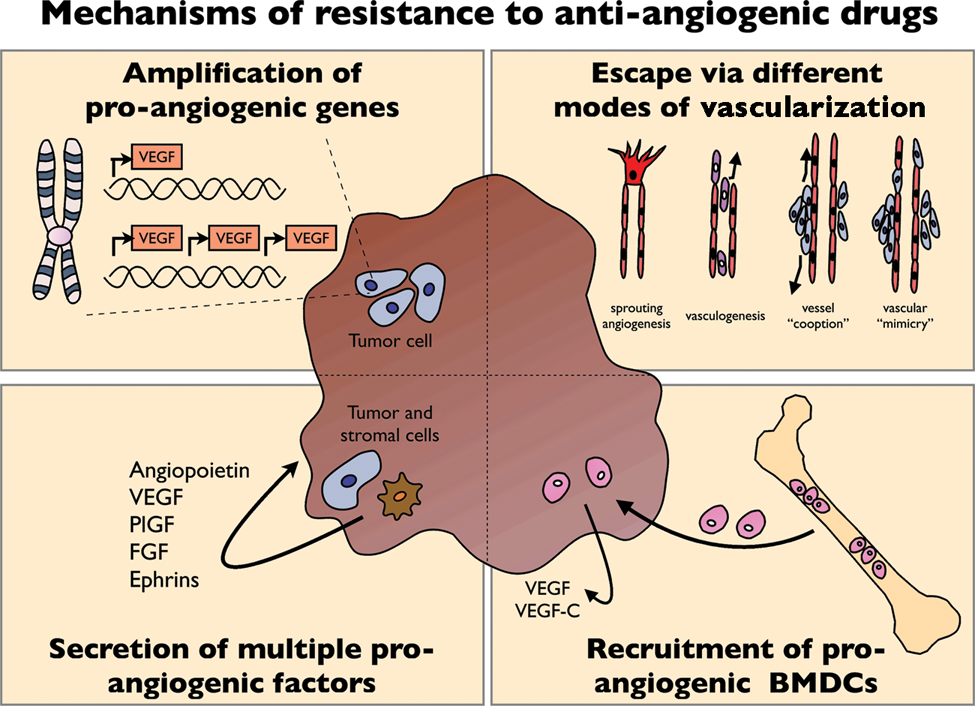
Principles of resistance against anti-angiogenic treatment.
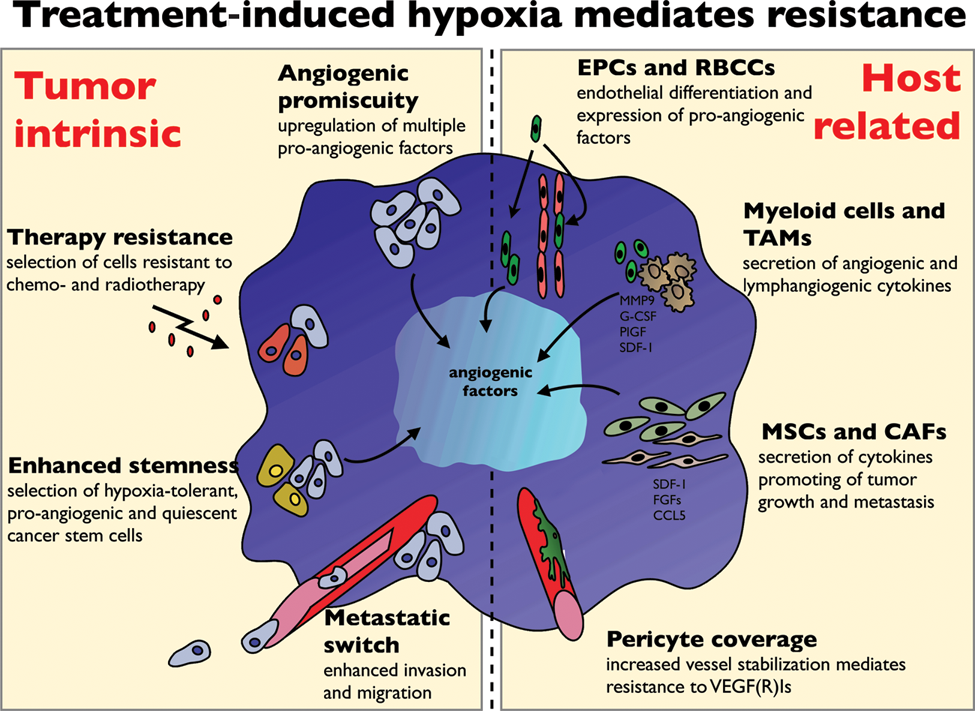
Treatment-induced hypoxia mediates resistance against anti-angiogenic therapy at the interface between tumor and host. Hypoxia-induced mechanisms of tumor cell escape
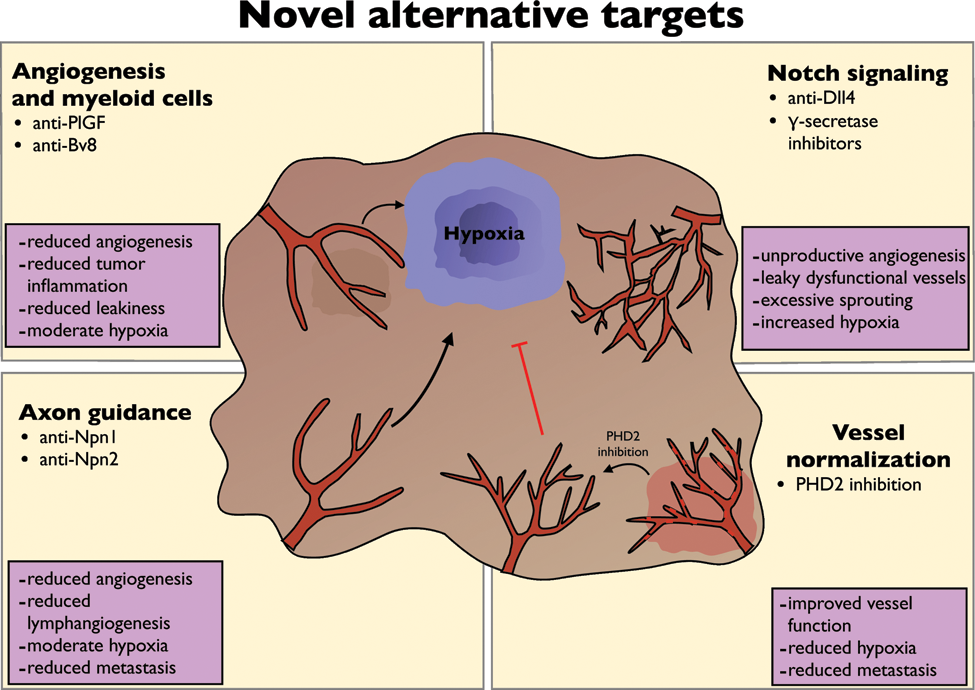
Alternative targets to overcome resistance.
Role of Malignant Cells in Anti-Angiogenic Escape
Malignant cells contribute to resistance against anti-angiogenic therapy via several mechanisms. A first mechanism relates to the promiscuity of tumors to produce angiogenic signals (Figure 1). Indeed, murine and human tumors express a myriad of angiogenic factors that can substitute for one another. 35-37 Such molecules are released in increasing amounts during tumor progression. 38,39 Most likely, different tumors exploit changing combinatorial codes of angiogenic factors at consecutive stages of tumor progression or metastasis that are, in turn, changed again by the type and stage of treatment. Future translational and biomarker studies are needed to crack these angiogenic codes. A challenge is, however, that expression of an angiogenic cytokine may correlate with disease stage and progression and act as a reliable prognostic biomarker but not necessarily be a suitable anti-angiogenic drug target or predictive biomarker of the response to anti-angiogenic agents. 40,41
Anti-angiogenic therapy itself may also evoke escape mechanisms that rescue tumor vascularization. For instance, VEGFR-2 blockade in tumor-bearing mice upregulates expression of placental growth factor (PlGF), VEGF, angiopoietin-1, fibroblast growth factor (FGF) family members, and other angiogenic factors. 35,36 Also, in glioblastoma patients, plasma levels of FGF-2 and stromal-derived factor 1 (SDF-1) are increased upon disease progression under VEGF-targeted therapy, 25 whereas PlGF and VEGF are also upregulated in colorectal and renal cancer patients after treatment with VEGF(R)Is. 42,43 Release of such angiogenic factors can be induced by hypoxia in the tumor tissue, caused by vessel pruning, but induction of these and other angiogenic cytokines, such as granulocyte colony-stimulating factor (G-CSF) and SDF-1, has also been attributed to production by healthy tissues. 35,36,44,45 A possible implication (and risk) of this iatrogenic induction of angiogenic signals is that they may cause angiogenesis to flair up, in a rebound, upon sudden arrest of VEGF-targeted therapy. This indeed has been observed in preclinical tumor models 28 as well as in cancer patients. 25 Such findings, together with clinical results that bevacizumab prolongs survival when continued beyond disease progression, 8 argue in favor of anti-angiogenic maintenance therapy, although further clinical confirmation would be required.
Recently, VEGF inhibitors were shown to promote tumor cell invasiveness and metastasis in several preclinical cancer models. 45,46 These data corroborate earlier preclinical findings of enhanced invasiveness of glioblastoma after VEGF blockade. 47 Emerging clinical evidence also reveals a higher incidence of glioblastoma relapse at distant sites after treatment with bevacizumab and chemotherapy than chemotherapy alone. 48 Various mechanisms may explain why VEGF-targeted therapy promotes such a shift to increased malignancy. 49 First, the low oxygen levels in tumors, regularly approaching anoxic conditions, represent a hostile microenvironment from which tumor cells try to escape to distant metastatic sites. 50-52 Besides hypoxia, VEGF(R)Is also induce the release of a “cytokine storm” by healthy tissues (see above), which induces a systemic “pseudo-inflamed” state that may promote tumor extravasation, metastatic lodging, and recruitment of angio-competent myeloid cells to primary tumors or premetastatic niches 35,36,44,45,50,53,54 (Figure 2). The clinical implications of these experimental observations need to be further evaluated, as it remains to be determined whether the aforementioned phenomena occur at all in cancer patients and might be related to the dose of anti-angiogenic regimen administered. Nonetheless, these findings call into question whether depriving tumor cells from their vascular supply as maximally as possible is desirable or, instead, should be balanced to prevent tumor invasiveness and metastasis, especially in patients with early stage, curable, nonmetastatic disease.
Although certain tumor cells try to escape from the hostile hypoxic environment, others try to become more hypoxia tolerant. 55-58 Notably, cancer stem cells (CSCs), which might contribute to the recurrence of metastasizing cancers in humans, home into hypoxic tumor regions, where they can sustain self-renewal. 59 However, other studies document that CSCs are also present in perivascular niches, 60 release angiogenic factors in hypoxic conditions, and establish a permissive vascular niche. 61 Consistent herewith, anti-angiogenic agents deplete CSCs from tumors in preclinical models. 60 It is unclear whether CSC subpopulations exist that differ in their addiction or tolerance to hypoxia.
Role of Endothelial (Progenitor) Cells in Anti-Angiogenic Escape
Another reason why tumors may become refractory to VEGF-targeted therapy is that they form vessels via different processes, which can partially substitute for one another. Indeed, certain tumors become vascularized via sprouting angiogenesis (growth of new branches), whereas others co-opt existing vessels and form vessels through intussusception (growth within itself), vasculogenic mimicry (via dedifferentiation of tumor cells), or postnatal vasculogenesis (via differentiation of bone marrow–derived or cancer stem cells). 62 Even though the molecular basis (and the relative role of VEGF) in all these distinct modes of vessel growth remains incompletely understood, one can intuitively assume that a single anti-angiogenic agent may not be capable of neutralizing all these nonoverlapping mechanisms of vessel growth.
The molecular mechanisms and role of VEGF in tumor vessel sprouting have been most intensively studied. Less well characterized are the other processes of vessel growth, whereby tumors secure their vascular supply; given space restraints, we restrict our discussion here to only some of those mechanisms. Co-option of preexisting blood vessels has been documented in various tumors, 63,64 including rat glioblastomas, 63 human pancreatic carcinomas, 65 non–small cell lung cancers (NSCLCs), and pulmonary metastases. 64 The molecular basis of tumor vessel co-option remains largely enigmatic, although an interplay of angiopoietins and VEGF has been implicated. 63 An outstanding question is whether vessel co-option may explain in part why bevacizumab is largely ineffective in pancreatic cancer 26 and why only a third of NSCLC patients respond to bevacizumab. 6
Vascularization of tumors via vasculogenesis occurs to a variable extent in some but not in all tumors, but overall, the relevance and precise mechanisms of this mode of tumor vessel growth remain debated. 66-68 Circulating endothelial progenitor cells (EPCs) can exert a bystander role in neovascularization by secreting angiogenic cytokines or by differentiating to endothelial cells into nascent vessels (vasculogenesis). 68 Consistent with their role in tissue regeneration, EPCs are also recruited when the tumor vasculature is destroyed by chemo/radiation therapy, both in preclinical models as well as in clinical studies. 69-71 Experimental and clinical studies further indicate that circulating EPC levels correlate with tumor size. 72,73 VEGF and other factors recruit these EPCs, explaining why VEGF inhibitors reduce EPC mobilization in cancer patients 25,43,74 and why EPC levels rebound in glioblastoma patients during drug-free intervals of a pan-VEGF inhibitor. 25 In breast cancer patients during drug-free intervals after chemotherapy, EPCs increase similarly, concomitant with a rise of plasma VEGF and other pro-angiogenic cytokines. 70 EPCs have also been implicated in conferring resistance against vascular-disrupting agents and certain types of chemotherapy in preclinical cancer models. 71,75
Not only the number but also the function of tumor vessels determines the overall response to anticancer treatment. Malignant cells release excessive amounts of VEGF and other angiogenic factors, not only because they are deprived of oxygen but also because oncogenes activate their gene transcription. Perivascular myeloid cells also participate in flooding tumor vessels with VEGF (see below). 14,53,76 As a result, these angiogenic signals start to act as “abnormalization factors,” which convert the tumor vasculature into a complex chaotic network of abnormal tortuous vessels that are leaky and lined by a highly disorganized endothelium 14,77,78 ; all these changes render tumor vessels dysfunctional, impair perfusion, and increase tumor hypoxia. 14 By partially normalizing these abnormal tumor vessels, VEGF inhibitors improve perfusion and drug delivery in some 79,80 but not in all preclinical studies. 81 Normalization of tumor vessels may also improve tumor oxygenation and responsiveness to irradiation in preclinical models 14,82 and can explain why bevacizumab amplifies the activity of cytotoxic agents. 5-7 However, tumor vessel normalization may depend on the dose and type of anti-angiogenic agents. At high doses, VEGF inhibitors induce excessive vessel pruning rather than normalization, which impairs perfusion, thereby creating a hostile hypoxic microenvironment from which tumor cells try to escape (see above). Anti-abnormalization strategies may offer novel opportunities to reduce anticancer resistance (see below).
It was originally postulated that endothelial cells, unlike malignant cells, are genomically stable and would therefore not be capable of escaping anti-angiogenic therapy. Although this is true in most cases, occasional studies report that endothelial cells in experimental and human tumors may harbor genetic abnormalities. 83,84 In addition, a recent report documented that murine tumor endothelial cells may have stem cell–like properties and can differentiate to other cell types. 85 An outstanding question is therefore whether such multipotent or genomically unstable endothelial cells might react differently or respond less to VEGFR(I) therapy and contribute to evasive escape from such therapy.
Role of Pericytes and Smooth Muscle Cells
Pericytes and smooth muscle cells promote maturation and stabilization of vessels and hence can influence the responsiveness of tumor vessels to anti-angiogenic therapy. By releasing platelet-derived growth factor–B (PDGF-B), endothelial cells recruit pericytes and render nascent vessels more stable, quiescent, and mature. 86 Overexpression of PDGF-B in experimental tumors increases pericyte coverage of tumor vessels and reduces tumor growth because pericytes inhibit endothelial proliferation and thereby reduce vessel density. 87 This may explain why immature, “naked” growing blood vessels are more susceptible to VEGF-targeted agents, 88 whereas mature, pericyte-covered vessels persist. 29,89,90 The cross-talk between VEGF and PDGF-B may also explain why combinations of VEGF and PDGF receptor (PDGFR) inhibitors are more effective in pruning tumor vessels than VEGF inhibitors alone. 89,90
However, recent studies call the strategy to skin tumor vessels from their mural cell coat into question. Indeed, not all (pre)clinical studies document that VEGF/PDGFR combination therapy is more effective in blocking tumor growth. 91,92 Furthermore, because pericytes impede tumor cell intravasation, caution is warranted to block pericyte recruitment indiscriminatively, as such strategies may fuel metastasis. 93 Also, by normalizing leaky and tortuous tumor vessels, vessel maturation through coverage of endothelial cells by pericytes may improve drug anticancer delivery. Because VEGF inhibits PDGFRβ-dependent recruitment of pericytes around immature vessels, 94 VEGF blockers improve the normalization of tumor vessels. 14 Last, loss of pericyte coverage can enhance vessel leakage, which might promote formation of ascites or edema. 95 Thus, the precise clinical consequences of pericyte-targeted therapy require further study.
Role of Cancer-Associated Fibroblasts
Cancer-associated fibroblasts (CAFs) create a microenvironment that promotes tumor growth, angiogenesis, and metastasis. 96-98 The degree of their infiltration correlates with poor outcome in cancer patients. 96 Besides stimulating angiogenesis by producing angiogenic cytokines, CAFs also promote experimental tumor vasculogenesis via secretion of SDF-1, which mobilizes and recruits bone marrow–derived EPCs in a VEGF-independent manner. 98 Furthermore, CAFs of VEGF(R)I-resistant tumors upregulate PDGF-C, which rescues vascularization of these tumors. 99 In addition, by increasing the coverage of experimental glioma vessels with pericytes, PDGF-C counteracts the vessel pruning effect of VEGFR-2 blockade. 100 Hence, neutralization of PDGF-C enhances the effect of VEGF(R)Is. 99
Mesenchymal stem cells (MSCs) are bone marrow progenitors that contribute to tissue regeneration. 101 Like CAFs, these cells are recruited via VEGF-independent mechanisms to experimental gliomas and colon and breast cancers, 97,102,103 where they stimulate angiogenesis by secretion of VEGF and other angiogenic cytokines. 104 MSCs also promote metastasis by secreting the chemokine RANTES, which stimulates tumor cell invasiveness. 97 Whether CAFs and MSCs participate in the evasive escape from VEGF-targeted therapy in cancer patients is currently unknown.
Myeloid Cells Contribute to the Resistance against VEGF(R)Is
Role of CD11b+Gr1+ Myelo-Monocytic Cells and TAMs
Gr1+CD11b+ cells comprise a heterogeneous population of myeloid cells that are recruited from the bone marrow to tumors, where they confer resistance to anti-angiogenic treatment in experimental tumor models. 105,106 G-CSF, produced by tumors, induces mobilization and recruitment of these myeloid cells. 105,107 Notably, VEGF(R)I-refractory tumors contain more Gr1+CD11b+ cells than their VEGF(R)I-sensitive counterparts; furthermore, sensitive tumor cells become resistant when admixed with Gr1+CD11b+ cells, isolated from resistant mice, because Gr1+CD11b+ cells release angiogenic factors. 105,108 Intriguingly, the population of Gr1+CD11b+ cells also contains myeloid-derived suppressor cells (MDSCs), which promote tumor growth by inhibiting the antitumoral immune response 109 and have been detected in renal cancer patients. 110 Whether MDSCs mediate resistance to anti-angiogenic medicine warrants further study.
Macrophages acquire different phenotypes: the M1 type has been associated with microbial killing, whereas the M2 type stimulates angiogenesis, lymphangiogenesis, invasion, and immunosuppression 111,112 and is associated with tumor progression, metastasis, and poor prognosis in cancer patients. 53,113 Tumor-associated macrophages (TAMs) belong predominantly to the M2 phenotype, but tumors also contain M1 polarized macrophages. 111,112 TAMs mediate in part the resistance of murine tumors against VEGF(R)Is because macrophage depletion enhances their responsiveness to different VEGF targeting agents. 36 Furthermore, blockade of PlGF, which chemoattracts TAMs, by a monoclonal antibody (αPlGF) blocks their recruitment to murine tumors, leading to growth inhibition of VEGF(R)I-resistant cancers. 36 Because PlGF is upregulated upon VEGF(R)I treatment, increased recruitment of pro-angiogenic and resistance-conferring TAMs by PlGF might be a mechanism of tumors to compensate for vessel pruning after anti-angiogenic therapy and contribute to resistance.
Another subtype of myeloid cells (i.e., the Tie2-expressing monocytes [TEMs]) is scarce in healthy tissue but present in human solid tumors. 114,115 TEMs are recruited to human xenograft tumors by angiopoietin-2 (Ang2), 115 which is upregulated in hypoxic tumors in response to anti-angiogenic treatment in some but not in all preclinical studies. 35,36,53,116 TEMs stimulate tumor angiogenesis by releasing bFGF 114 and by downregulating the anti-angiogenic cytokine IL-12. 117 Whether TEMs promote escape from anti-angiogenic treatment in human cancer remains to be determined.
A recent mouse genetic study reported that absence of VEGF in perivascular myeloid cells promotes tumor growth and progression but at the same time also increases tumor sensitivity to chemotherapy. 76 Notably, tumor vessel numbers were not different, but tumor vessel function was improved because of normalization. These vascular changes reduced tumor hypoxia, which can explain why tumor cells proliferated more and responded better to chemotherapy. 118 It will be interesting to await the effects on metastasis in this model, as this process is usually promoted by tumor hypoxia. 50,51 Regardless, these findings highlight the importance of VEGF, produced by perivascular myeloid cells, in driving the tumor vessel abnormalization switch. These results might also provide an additional explanation or at least raise the question why VEGFR(I)s are often more effective in inhibiting tumor growth when administered together with chemotherapy.
Role of Pro-Angiogenic VEGFR-1+ Myeloid Cells
Different subpopulations of bone marrow–derived CD45+ myelomonocytic cells promote angiogenesis via secretion of angiogenic factors in preclinical cancer models. 53 For example, recruited bone marrow–derived circulating cells (RBCCs), expressing CD11b, CXCR4, VEGFR-1, and CX3CR1, are lured toward VEGF and trapped around vessels, where they promote proliferation of resident endothelial cells, in part via secretion of MMP9 that liberates VEGF. 119 The perivascular retention of these RBCCs is dependent on the production of the CXCR4 ligand SDF-1 by pericytes and fibroblasts. 119 Interestingly, this cytokine is upregulated in tumors during anti-angiogenic escape, raising the question whether it might promote retention of pro-angiogenic RBCCs and contribute to VEGF-independent angiogenesis. 35,36,119
Third-Generation Anti-Angiogenic Strategies
Anti-Myeloangiogenic Strategies
Resistance-conferring CD11b+Gr1+ myeloid cells upregulate the pro-angiogenic protein Bv8 in response to G-CSF, which enhances their recruitment (see above). Bv8 levels are elevated in tumors that develop resistance to VEGF inhibition, possibly due to enhanced recruitment of myeloid cells to hypoxic tumors. 106 Anti-Bv8 antibodies (αBv8) not only inhibit growth and angiogenesis of VEGF(R)I-resistant tumors but also enhance the anticancer activity of VEGF inhibitors. 106 In the RIP-Tag model of pancreatic islet carcinogenesis, αBv8 is effective in prevention but not in therapeutic inhibition of established tumors. 120 Because Bv8 expression is downregulated in hepatocellular carcinoma as compared to normal liver without correlation between expression levels of Bv8 and angiogenesis, further studies on the role of Bv8 in human cancer are warranted. 121
PlGF has been implicated in the resistance against anti-VEGF therapy. 122 Its levels correlate with clinical outcome in several, though not all human cancers. 122 Furthermore, in cancer patients, PlGF is upregulated upon treatment with VEGF inhibitors. 42,74 Overexpression of PlGF can stimulate or inhibit tumor growth, depending on whether its levels are, respectively, physiological or supra-physiological, that then reduce the amount of angiogenic VEGF homodimers. A monoclonal anti-PlGF antibody (αPlGF) inhibits tumor angiogenesis and growth, reduces the recruitment of resistance-conferring macrophages, and has direct cytostatic effects on tumor cells. 36 Which tumors are sensitive or resistant remains to be further determined. In comparison to VEGF(R)Is, treatment with αPlGF causes less hypoxia or a less prominent pro-angiogenic escape profile. 36 Also, αPlGF enhances the anticancer activity of VEGF(R)Is and has an attractive safety profile. 36 The therapeutic potential of a humanized version of αPlGF (TB403) is currently being evaluated in cancer patients.
Promoting Nonproductive Angiogenesis
Starving tumors from oxygen and nutrients by pruning their blood vessels has long been a goal of anti-angiogenic therapy. This dogma has been recently challenged by preclinical findings that a monoclonal antibody against the Notch-ligand Delta-like 4 (αDll4), which stimulates—not inhibits—the growth of supernumerary vessels, suppressed rather than promoted tumor growth. 123-125 This apparent paradox can be explained by the fact that these vessels are functionally crippled and hypoperfused, a process coined “nonproductive angiogenesis.” 123-125 αDll4 inhibits the growth of VEGF(R)I-sensitive and -resistant cancer models. 123,124 In humans, Dll4 is upregulated by the vasculature of renal cell carcinoma and bladder cancer. 126 Because Dll4-Notch blockade impairs tumor perfusion and oxygenation, its effects on drug delivery and metastasis and the induction of rescue vascularization or inflammatory cell recruitment remain to be evaluated. 24,49,55
Inhibiting Vessel Maturation or Lymphangiogenesis by Blocking Neuropilins
Several molecules, originally discovered as axon guidance cues, also regulate angiogenesis. 127,128 Neuropilins (NRP1-2) regulate angiogenesis as coreceptors for VEGF and PlGF 129 or as signaling molecules independently of VEGF receptors. 130 They are expressed on different types of tumor cells, 131 and tumor NRP1 levels correlate with advanced disease and prognosis in certain cancers, such as prostate and lung cancer. 132-134 A preclinical study indicates that a monoclonal antibody blocking the binding of VEGF to NRP1 (αNRP1VEGF) inhibits tumor growth. Upon combination with anti-VEGF, tumor growth was inhibited more, 131,135 raising the question whether αNRP1VEGF may perhaps inhibit other ligands, such as PlGF, HGF, FGFs, and so on. Complementary to anti-VEGF, αNRP1VEGF inhibits pericyte coverage, thereby rendering tumor vessels more sensitive to VEGF inhibitors. 89,135
NRP2 is overexpressed on lymphatics in tumors. 136 An anti-NRP2 antibody (αNRP2) inhibits tumor lymphangiogenesis and metastasis via blocking interactions of NRP2 and VEGF-C, without influencing primary tumor growth or angiogenesis. 136 Interestingly, NRP2 is also expressed by the tumor vasculature, and its specific ligand Semaphorin 3F was shown to act as a chemorepulsant for endothelial cells. 137,138 Indeed, tumoral Sema3F expression induces a poorly vascularized and nonmetastatic phenotype, raising the possibility that Sema3F might constitute a novel angiogenesis inhibitor and antimetastatic agent. 137,138
Antivessel Abnormalization Strategies
Conventional anti-angiogenic strategies aim to inhibit neovascular growth and prune existing vessels, but they bear the risk of inducing tumor hypoxia with the risk of enhanced tumor invasiveness and metastasis and reduced tumor response to chemo/radiotherapy. 49,55 Recent genetic studies in mice reveal that targeting tumor vessel function, rather than their numbers, may provide an alternative to reduce tumor malignancy, invasiveness, and metastasis. 78 An ancestral function of endothelial cells is to provide optimal perfusion of blood vessels to secure tissue oxygenation. Endothelial cells therefore are equipped with the oxygen sensor PHD2, which senses shortage of oxygen and mounts a feedback loop to readjust vessel perfusion and oxygenation. 78 Tumor vessels are highly abnormal, not only because they are more tortuous but also because their pseudo-stratified multilayered endothelial cell layer is highly disorganized and leaky, with endothelial cells protruding intraluminal cell extensions that obstruct the lumen and impair perfusion and oxygen supply. 14,77,78
Partial loss of PHD2 induces endothelial cell normalization, resulting in a streamlined phalanx formation (comparable to the ancient formation of Greek soldiers) of quiescent, tightly interconnected, and orderly arranged endothelial cells, which improves perfusion and oxygenation. As a result, tumors in PHD2 haplodeficient mice invade the surrounding tissue less and also metastasize less. Thus, blocking PHD2 in endothelial cells may offer a conceptually novel strategy to convert tumors to a more benign phenotype, by improving—not impairing—tumor vessel perfusion. However, the role of PHD2 in tumor biology is complex, as this oxygen sensor might regulate different oncogenic versus tumor-suppressive mechanisms in a dose-dependent manner. 139 The role of PHD2 in human cancer is largely unknown, but one study documented a correlation of intratumoral PHD2 levels with a more aggressive phenotype of human head-and-neck cancer, which might provide a rationale to generally inhibit tumoral PHD2. 140 In any case, further study is required to develop specific PHD2 inhibitors for clinical use in the future.
Conclusion
Four years after approval of the first anti-angiogenic drug, primary or acquired resistance remains an important problem to increase efficacy. Very recently, exciting novel concepts have emerged from preclinical research, which may start to explain in part the resistance to anti-angiogenic agents in the clinic. These insights provoke several outstanding questions with regard to their relevance in cancer patients: Can VEGF inhibitors also fuel metastasis in cancer patients? Do human tumors switch mechanisms to secure their vascular supply during anti-angiogenic treatment? How relevant is the role of EPCs in mediating resistance? What are the consequences of therapies targeting pericytes? Do CAFs, MSCs, and different populations of myeloid cells confer resistance, and if yes, how? In the meantime, several promising third-generation drug candidates with potential to induce less or overcome resistance have emerged. The field is facing the challenge to translate drug candidates such as αBv8, αPlGF, Notch inhibitors, αNpn1-2, and vessel normalization strategies into the clinic; develop strategies to enhance efficacy of established drugs; and validate faithful biomarkers to monitor anti-angiogenic treatments. But with increasing understanding of mechanisms of blood growth and functionality, hope is rising that these goals can be reached in the future.
Footnotes
P. Carmeliet is named as inventor on patents, claiming subject matter that is partially based on the results described in this article. The patents are licensed/submitted, which may result in royalty payment to Peter Carmeliet.
S.L. is supported by Deutsche Krebshilfe, T.S. is supported by Deutsche Forschungsgemeinschaft, and P.C. receives “Long-term structural funding: Methusalem funding by the Flemish Government” and support through the Fund for Scientific Research–Flemish Government (FWO; G0125.00 and G.0121.02), the European Union (QLRT-2001-0195), Concerted Research Activities–Katholieke Universiteit Leuven (GOA2001/09 and GOA/2006/11), and the Belgian Science Policy (IUAP-P5/02 and IUAP-P6/30).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.