
Abstract
Anti-NMDA Receptor (NMDAR) Encephalitis (NMDARE) is an autoimmune disorder that is often debilitating and difficult to diagnose. Patients, especially those with underlying neuropsychiatric disorders, may experience delayed or misdiagnosis of NMDARE. Here, we report on a patient with known congenital leukodystrophy (CLD) and epilepsy with a challenging diagnosis of NMDARE. The patient first presented with progressive behavior changes and seizure-like episodes. Initial workup, including video EEG and brain MRI, were mostly unremarkable, and the patient's symptoms were resistant to treatment with multiple anti-epileptic drugs. Given the patient's complicated clinical history, his presentation was initially thought of as progression or exacerbation of his chronic disease. With continued lack of improvement, autoimmune encephalitis was considered. The patient was started on immunotherapy and autoimmune encephalitis panels were sent, which came back positive. He continued to improve over the next weeks and months. Despite a growing body of literature, our knowledge on confirmed risk factors for NMDAR remains limited outside of young age, ovarian teratomas, and herpes encephalitis. We know that maintenance of the blood brain barrier is key to preventing autoimmune disorders of the central nervous system (CNS), and multiple congenital leukodystrophies exhibit pathology in the neurovascular unit. This is the first described case of anti-NMDA receptor encephalitis in a patient with an underlying congenital leukodystrophy, which may reflect an underreported NMDAR encephalitis risk factor. With limited known risk factors and time to diagnosis and treatment so important, this case may reflect an important and underreported risk factor for NMDAR.
Keywords
Introduction
Anti-NMDA Receptor (NMDAR) Encephalitis (NMDARE) is an autoimmune disorder that is often debilitating and difficult to acutely diagnose. Presenting symptoms vary widely between patients, and risk factors for the disease are largely unknown. Patients, especially those with underlying neuropsychiatric disorders, may experience delayed or misdiagnosis of NMDARE. Here, we report on a patient with known congenital leukodystrophy (CLD) and epilepsy with a challenging diagnosis of NMDARE. This is the first described case of anti-NMDA receptor encephalitis in a patient with an underlying congenital leukodystrophy which may reflect an underreported NMDAR encephalitis risk factor.
Case Description
A 25-year-old man presented to the emergency department with uncharacteristic agitation and seizure-like episodes. His medical history included congenital leukodystrophy associated with mild cognitive impairment and epilepsy. He was seizure free since prior to 2010 on Carbamazepine 200 mg BID. In 2018, the patient discontinued all antiseizure medications (ASMs), remaining seizure free off ASMs until this presentation in 2021. At baseline he ambulated independently, was active despite mild spasticity, and had moderate dysarthria and limited vocabulary but was able to communicate fully. In addition, he maintained gainful employment.
His previous seizure semiology included focal seizures and left side shaking, and was well controlled on carbamazepine monotherapy. Work up for cause of leukodystrophy was unrevealing including a 700 gene panel and assays for N-acetylaspartate (NAA), aryl sulfatase, very long chain fatty acids (VLCFA), phytanic acid, B galactocerebrosidase, and oligosaccharide disorders.
In the weeks prior to admission, the patient developed new agitation and confrontational behavior. Starting 1 week prior, he had seizure-like activity with stiffening, rhythmic right arm and hand movements, and speech arrest. He exhibited other abnormal movements while awake including snapping and pinching his nose.
He presented to the emergency department due to worsening seizure frequency. Labs were negative for infections and metabolic disturbances. Brain MRI without contrast was stable compared to a remote prior study, showing prominent periventricular white matter disease (Figure 1). He was unable to tolerate repeat studies with contrast due to controlled dyskinetic movements, and risk of sedation was felt to be too great by family. Continuous video EEG showed electroclinical seizures of left frontal onset correlating with right arm movements. Due to his past medical history and unclear baseline level of function provided by the family, the patient was initially treated for breakthrough epilepsy with escalation of AEDs (carbamazepine, levetiracetam, and lacosamide) without termination of seizures. He required a percutaneous endogastric (PEG) tube for nutrition. Select images from MRI brain without contrast obtained on hospital day 1 of presenting admission. (A) Axial T2-weighted FLAIR image demonstrates marked confluent periventricular white matter signal abnormality and volume loss. (B) Coronal T2 weighted image demonstrates similar features. When compared to CT head from 1 year prior and MRI brain from 16 years prior, there is no significant change. No abnormalities of the hippocampi, such as atrophy or T2 hyperintensity, were noted.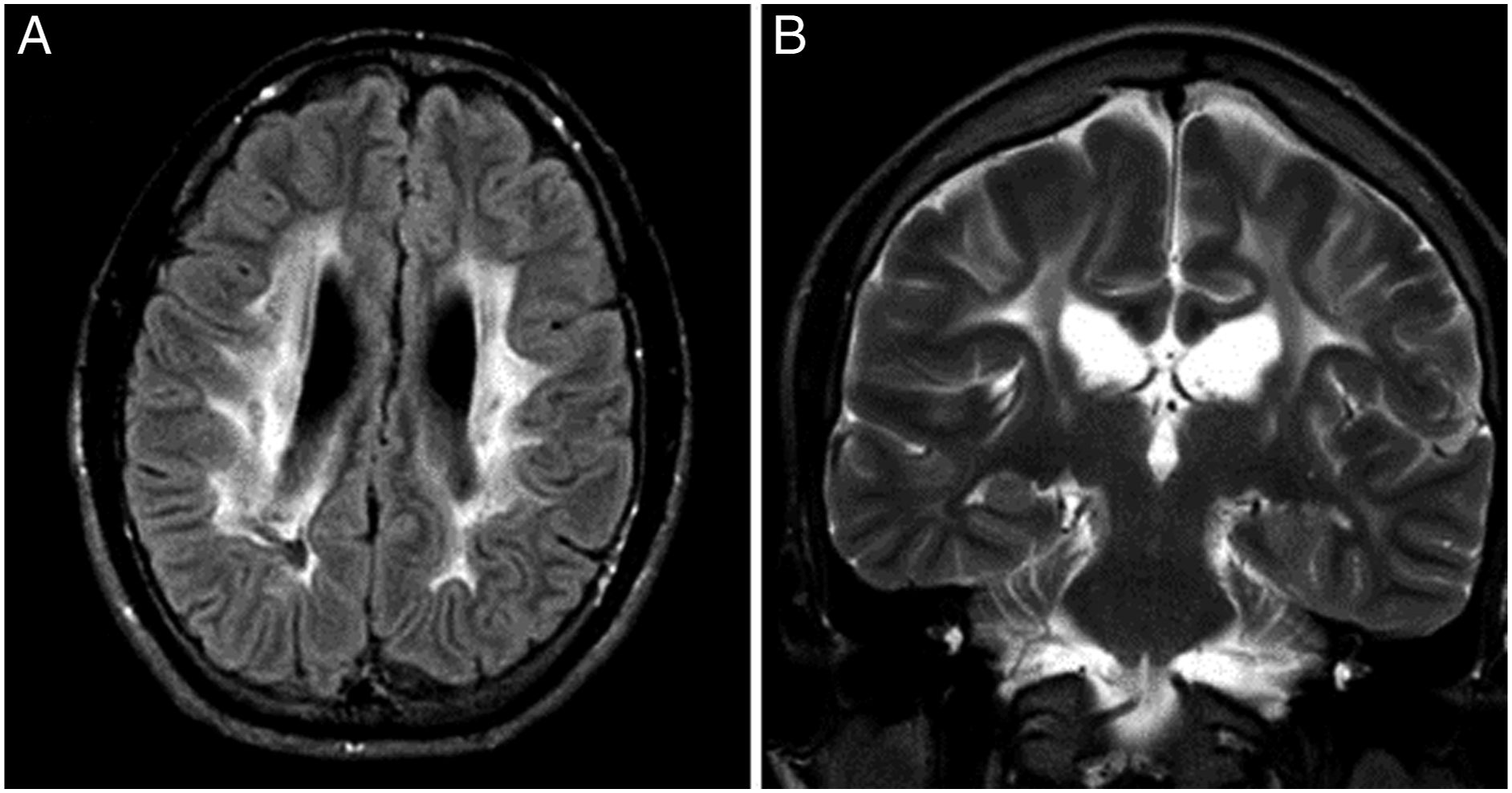
The medical team reapproached the differential diagnosis of refractory seizures by having another discussion with the patient’s father. This revealed an accurate functional baseline and confirmed that the behaviors and seizures were completely new symptoms for this patient. A workup was initiated for alternative causes of new onset seizures, including a lumbar puncture (cerebrospinal fluid (CSF) with normal cell count, glucose, protein but slightly elevated opening pressure at 30 cm H2O). Serum and CSF samples were sent for “Mayo Clinic ENS2/Encephalopathy, Autoimmune/Paraneoplastic Evaluation” and “Mayo Clinic ENC2/Encephalopathy, Autoimmune/Paraneoplastic Evaluation” panel testing, respectively. These panels test for a range of antibodies used to aid in diagnosis of new onset encephalopathy workup.
There was high concern for autoimmune encephalitis at this time, in part because of an APE2 score of 8 and negative workup for other causes. APE2 or antibody prevalence in epilepsy and encephalopathy, scoring can be used in patients with seizures or who are encephalopathic to predict antineuronal antibody testing. A score of 4, or greater, is generally regarded as a strong indicator of neural-specific antibodies. 1 Thus, appropriate treatment was initiated while serum and CSF panels were pending. A 5-day course of empiric IVIG was begun. He had moderate improvement in responsiveness, alertness and communication beginning soon after his fifth day of IVIG; he was able to watch movies on a tablet and follow simple commands. Dyskinetic movements, clonus, and rigidity, however, were largely unchanged, and cognitive status was still far from baseline. 10 days after his first IVIG infusion, the ENS2 and ENC2 panels resulted. Serum panel testing resulted positive only for anti-NMDA receptor IgG antibodies and, similarly, the CSF panel was positive for only the NMDA receptor antibody cell-binding assay. Rituximab was adminitersted at this time, given confirmation of an NMDA-receptor encephalitis (NMDARE) diagnosis by the above. He had progressive improvement in the above abilities and symptoms over the next 10 days, at which point he was deemed appropriate for discharge.
He received first line immunotherapy with methylprednisolone and intravenous immunoglobulin (IVIG) with partial improvement. Therapy was escalated to rituximab. Within 2 weeks, his alertness and attention improved with resolution of his abnormal orofacial movements, rhythmic movements, and staring spells. At follow-up 5 weeks later, he was at his cognitive baseline and complained only of mild physical deconditioning, with the neurological exam showing no new deficits. He was tapered to lacosamide monotherapy and PEG tube was removed over the following weeks with no return of neuropsychiatric symptoms nor seizures.
Discussion
First discovered in 2007, NMDARE is a complicated neuropsychiatric immune-mediated disease. While it was first thought to be highly associated with ovarian teratomas, our understanding and knowledge has progressed immensely in the years since. Symptom onset often has a post-viral prodromal phase, with the more typical and severe symptoms developing in the coming weeks to months. Because of the wide range of signs and symptoms it can take on, diagnosis of the disease can be challenging, even in those without underlying neuropsychiatric conditions. The median time to diagnosis of NMDARE is 4-8 weeks with longer diagnoses occurring in older patients, presumably because they do not fit the expected NMDARE risk factor of young age. 2 While survivors of the disease can make a near full recovery, mortality is estimated to be upwards of 5%. Given that time to treatment is a highly significant independent risk factor for patient outcomes, identification of risk factors is critically important to the future of NMDARE diagnosis and care. 2
Despite a growing body of literature, our knowledge on other risk factors for NMDAR remains limited. Young age, ovarian teratomas, and herpes encephalitis are well established risk factors for NMDARE. 3 Demyelinating disorders including multiple sclerosis (MS) and neuromyelitis optica (NMO) have also been investigated as a risk factor with supportive case literature, with over 3.3% of patients in 1 NMDARE case series having comorbid MS. 4 However, on a population level, anti-NMDA antibodies are not increased among patients with demyelinating disease compared to healthy controls. 5 Autism is highly associated with CNS autoimmunity and case reports suggest that NMDARE may be an easily missed cause of regression, although autism itself has not been shown to be a risk factor for developing NDMAR. 6 Congenital disorders of the white matter however have not been investigated as a risk factor.
Maintenance of the blood brain barrier is key to preventing autoimmune disorders of the CNS. Studies suggest that serum NMDA antibodies are not uncommon at a population level (10.7% among healthy patients), but clinically a history of compromise to the blood brain barrier may determine if the antibodies lead to neuropsychiatric complication. 7 Multiple congenital leukodystrophies exhibit pathology in the neurovascular unit, even impairing the blood brain barrier significantly enough to correlate with contrast enhancement on CT scans.8,9 Impairment of the neurovascular unit and exposure of the brain to the peripheral immune system could represent a pathophysiologic link in this rare disease co-occurrence – but ultimately more case literature is needed to determine if CLD is a risk for NMDARE.
This case highlights the potential presenting symptoms and resultant added difficulty in diagnosis of NMDARE in a patient with an underlying CLD. As always, the importance of a thorough history, which in this case was initially difficult to obtain and possibly contributed to delayed diagnosis and treatment, cannot be overstated. While implicit bias garners the most attention in training curricula and has been shown to lead to delays in diagnoses, we must not forget that other types of bias exist. 10 In a patient with an underlying disease similar to the alternative and final diagnosis, as seen in our patient, we cannot fall into the trap of anchoring or other biases which can delay diagnosis and prognosis-altering treatment. This is the first reported case of NMDARE in a patient with CLD and a reminder that acute-subacute neurologic change, in patients with a congenital neurologic disorder, should always prompt consideration of a new pathology.
Conclusion
While acute neuropsychiatric disease in the previously neurologically healthy is more easily diagnosed as NMDA encephalitis, those with underlying disease such as CLD are likely at equal if not higher risk for the disorder. It is critically important to consider an early diagnosis of autoimmune encephalitis in patients with progression of underlying neurologic disorders with a careful, unbiased history and a thorough diagnostic work-up. Informed consent: Informed consent was obtained.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.