
Abstract
Non-alcoholic fatty liver disease (NAFLD) and atherosclerosis (AS) are two major phenotypes of metabolic syndrome that frequently coexist through a “liver–vascular axis” characterized by lipid dysregulation, chronic inflammation, and mitochondrial dysfunction. The liver kinase B1/AMP-activated protein kinase (LKB1/AMPK) signaling pathway, acting as a master regulator of energy and redox balance, has emerged as a central hub in this comorbidity. Activation of this pathway suppresses lipogenesis, enhances fatty acid oxidation, attenuates inflammation, and improves endothelial function, thereby interrupting the vicious metabolic–inflammatory cycle underlying NAFLD and AS. Natural products provide promising multi-target modulators of LKB1/AMPK. Polyphenols (such as curcumin, resveratrol, quercetin), terpenoids (such as nobiletin, betulinic acid), alkaloids (such as berberine), and glycosides (such as ginsenosides, salidroside) restore lipid homeostasis, induce autophagy, regulate oxidative stress, and modulate immune responses via LKB1/AMPK and its crosstalk with sirtuin 1 (SIRT1), peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α), and mammalian target of rapamycin (mTOR) signaling. Preclinical studies highlight their ability to improve hepatic steatosis, vascular inflammation, and plaque stability, underscoring their cross-organ therapeutic potential. However, pharmacological translation remains limited by poor bioavailability, variable pharmacokinetics, species differences, and inconsistent clinical outcomes. This review systematically summarizes comorbidity mechanisms of NAFLD and AS, regulatory roles of the LKB1/AMPK pathway, therapeutic actions of representative natural products, and the challenges ahead, aiming to guide precision strategies for metabolic syndromes.
Introduction
Non-alcoholic fatty liver disease (NAFLD) and atherosclerosis (AS) are two core manifestations of the metabolic syndrome that have risen to global prominence. Epidemiological data indicate that NAFLD affects roughly one quarter of the world's population and that AS-related cardiovascular disease remains the leading cause of mortality worldwide.1–3 Their frequent co-occurrence and reciprocal worsening amplify risks of major adverse cardiovascular events as well as progression to end-stage liver disease, making the NAFLD-AS axis a major clinical and research priority.4–6 This comorbidity calls for mechanistic frameworks that cross traditional organ boundaries rather than organ-centric approaches.
Mechanistically, NAFLD and AS share a network of interlocking pathologies-lipid dysregulation, chronic inflammation, oxidative stress, and insulin resistance (IR)-that drive reciprocal organ injury.7–9 Hepatic lipotoxicity promotes atherogenesis via enhanced secretion of very low-density lipoprotein (VLDL), formation of oxidized low-density lipoprotein (ox-LDL), and exosome-mediated suppression of macrophage cholesterol efflux (for example, by miR-30a-3p), while vascular dysfunction and impairment of high-density lipoprotein (HDL) in AS can feed back to aggravate hepatic lipid deposition and fibrogenesis.10–13 A growing body of imaging, biomarker, and molecular studies therefore supports the concept of a liver-vascular crosstalk in which the liver both signals to and is affected by the atherosclerotic milieu.
Among molecular hubs implicated in this crosstalk, the liver kinase B1/AMP-activated protein kinase (LKB1/AMPK) signaling axis emerges as a central integrator of energy, lipid, and immune homeostasis. LKB1/AMPK functions as a cellular energy sensor that directly phosphorylates targets such as acetyl-CoA carboxylase (ACC)-leading to reduced malonyl-CoA and enhanced carnitine palmitoyltransferase 1 (CPT1)-mediated fatty acid oxidation-while suppressing anabolic programs driven by sterol regulatory element-binding protein 1c (SREBP1c) and 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR). At the same time, it promotes vascular protection by enhancing endothelial nitric oxide synthase (eNOS) activity, inducing autophagy, and inhibiting nuclear factor kappa B (NF-κB).14–16 These direct AMPK actions are further modulated by secondary but highly relevant axes such as sirtuin 1 (SIRT1), mammalian target of rapamycin (mTOR), and nuclear factor erythroid 2-related factor 2 (Nrf2), which may act downstream of, in parallel with, or independently from AMPK to shape mitochondrial biogenesis, antioxidant defenses, and inflammatory set-points.17–19 Thus, targeting the LKB1/AMPK hub offers a rational strategy to concurrently ameliorate hepatic steatosis, systemic metabolic dysfunction, and atherogenesis.
Several reviews have previously addressed natural products and the LKB1/AMPK pathway in the context of NAFLD.20–22 However, these reviews primarily focused on liver-specific disease mechanisms and compound classes, with limited emphasis on vascular pathology or NAFLD–AS comorbidity. In contrast, the present review distinctively emphasizes the bidirectional liver–vascular crosstalk, highlights how natural products acting on LKB1/AMPK may simultaneously mitigate both hepatic and vascular lesions, and critically discusses pharmacological and translational challenges that remain obstacles to clinical application.
Natural products—including polyphenols, terpenoids, alkaloids, and glycosides—have attracted attention because many directly or indirectly activate LKB1/AMPK and simultaneously modulate complementary pathways such as SIRT1, Nrf2, and mTOR, yielding pleiotropic benefits in preclinical NAFLD and AS models (for example, curcumin, ginsenosides, berberine, resveratrol, and epigallocatechin gallate (EGCG)).23–25 However, a critical translational gap persists: most evidence is preclinical, and progress to the clinic is hampered by poor oral bioavailability, complex pharmacokinetics—including extensive first-pass metabolism and gut microbiota transformation—species-dependent differences, and inconsistent clinical trial outcomes. Addressing these pharmacological and delivery challenges is essential to convert promising mechanisms into effective therapies for patients.26–28
In this review we (1) synthesize epidemiological and mechanistic evidence linking NAFLD and AS, (2) summarize the biological functions of the LKB1/AMPK pathway that underlie this crosstalk, (3) review natural products that target this hub with emphasis on unique molecular mechanisms and available clinical evidence, and (4) critically discuss pharmacokinetic, safety and translational challenges that must be overcome to enable clinical application. By integrating mechanistic biology with translational realities, we aim to provide a focused roadmap for leveraging LKB1/AMPK-targeting natural products in the management of NAFLD-AS comorbidity.
Method
Literature Search and Selection Criteria for Natural Products
To identify natural products that modulate the LKB1/AMPK signaling pathway in the context of NAFLD, non-alcoholic steatohepatitis (NASH), or AS, a systematic literature search was conducted following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines. Five electronic databases—PubMed, Web of Science, Embase, Scopus, and the China National Knowledge Infrastructure—were searched from inception to June 30, 2025. Search terms combined pathway- and disease-related keywords (“LKB1” OR “serine/threonine kinase 11” OR “AMPK” OR “AMP-activated protein kinase”) with disease terms (“non-alcoholic fatty liver disease” OR “NAFLD” OR “non-alcoholic steatohepatitis” OR “NASH” OR “atherosclerosis” OR “AS”) and natural product-related terms (“natural product” OR “phytochemical” OR “herb*” OR specific compound names such as “curcumin”, “berberine”, “resveratrol”). References of retrieved articles and relevant reviews were manually screened for additional studies.
Inclusion and Exclusion Criteria
Eligible studies met the following criteria:
original experimental studies in cell culture (in vitro), animal models (in vivo), or clinical research on plant- or fungi-derived natural products (isolated compounds or defined extracts); direct experimental evidence of LKB1/AMPK pathway modulation (for example, changes in phosphorylated AMPK at Thr172, phosphorylated LKB1, or downstream targets), or mechanistic validation using AMPK/LKB1 inhibition, knockdown/knockout, or other perturbation experiments; and outcomes relevant to NAFLD, NASH, or AS (for example, lipid accumulation, inflammation, plaque area, endothelial function, liver enzymes).
Exclusion criteria comprised: review articles, editorials, and conference abstracts without full data; purely computational docking studies without experimental validation; synthetic derivatives without confirmed natural origin; studies lacking direct LKB1/AMPK readouts; non-peer-reviewed reports; and non-English or non-Chinese publications without accessible full text.
Data Extraction and Quality Assessment
Two reviewers independently screened titles, abstracts, and full texts, resolving disagreements by consensus. Extracted data included compound or extract identity, source, study model, dosing regimen, pathway evidence, key outcomes, and pharmacokinetic data where available. Risk of bias was assessed using the Systematic Review Centre for Laboratory animal Experimentation tool for animal studies and the Cochrane risk-of-bias tool for clinical trials. Mechanistic robustness was graded according to pre-specified criteria. The study selection process was documented in a Preferred Reporting Items for Systematic Reviews and Meta-Analyses flow diagram.
Pathological Mechanisms Linking NAFLD and AS
Association Between NAFLD and AS
Epidemiological and Biomarker Evidence
Multiple studies confirm a significant correlation between NAFLD and atherosclerotic cardiovascular risk, independent of confounders such as metabolic syndrome, smoking, and obesity.1–3 Patients with NAFLD exhibit higher carotid intima-media thickness (CIMT), coronary artery calcium (CAC) scores, and arterial stiffness (brachial-ankle pulse wave velocity; carotid-femoral pulse wave velocity) compared to non-NAFLD individuals.1–3,8,13 The severity of NAFLD demonstrates a dose-dependent relationship with AS risk: grade 3 hepatic steatosis correlates with significantly elevated CIMT, while advanced liver fibrosis is linked to increased CAC risk.4,5 Notably, even in young (<45 years) and non-obese individuals, NAFLD remains independently associated with subclinical AS.6–8
Beyond imaging-based indices, multiple biomarkers further support the NAFLD–AS link. Inflammatory and metabolic markers such as interleukin-6 (IL-6), C-reactive protein (CRP), Fetuin-A, and pentraxin-3 are elevated in NAFLD and correlate with AS progression, while low family with sequence similarity 19 member A5 levels show negative associations with hepatic steatosis and CIMT.14–16 Lipid-related markers including high triglyceride/high-density lipoprotein cholesterol (triglyceride [TG]/high-density lipoprotein cholesterol [HDL-C]) ratios, small extracellular vesicle microRNA-30a-3p, and ATP-binding cassette transporter (ABC)-A1/cholesterol 7 alpha-hydroxylase imbalance reflect cholesterol dysregulation in NAFLD–AS crosstalk.17–19 Molecular pathway markers such as receptor-interacting protein kinase 1, sirtuin 6 (SIRT6), and peroxisome proliferator-activated receptors indicate lipid–inflammation interaction, whereas chemokine (C-C motif) ligand 19 expression and CD4+ T-cell infiltration highlight immune involvement.17,20,21
Furthermore, clinical risk assessment tools—including non-invasive liver fibrosis scores (fibrosis-4 index; NAFLD fibrosis score) and imaging-based plaque characteristics (non-calcified plaque ratio)—hold predictive value for cardiovascular outcomes in patients with NAFLD.3,12,22 Together, these findings establish NAFLD as both a structural and molecular predictor of atherosclerotic burden.
Key Mechanisms
The pathological interplay between NAFLD and AS involves both direct effects of the LKB1/AMPK axis and secondary/synergistic pathways (eg, SIRT1; mTOR; Nrf2), which may act independently or downstream of AMPK activation.
Lipid Metabolism Dysregulation: NAFLD disrupts hepatic lipid metabolism, elevating TG, non-HDL-C, and ox-LDL while reducing HDL-C. This promotes VLDL secretion and cholesterol accumulation.23,24 Liver-derived exosomes (eg, miR-30a-3p) inhibit ABC-A1-mediated cholesterol efflux, driving macrophage foam cell formation and accelerating AS. Direct AMPK-mediated effects include upregulation of ABC-A1/ABC-G1 and scavenger receptor class B type I/lecithin-cholesterol acyltransferase to enhance HDL-dependent reverse cholesterol transport, 25 suppression of ox-LDL uptake, 26 and promotion of autophagy (FoxO3a–LC3/Unc-51 like autophagy activating kinase 1, ULK1 pathway). 27 Secondary mechanisms involve activation of SIRT1 (which independently deacetylates peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and p65) and inhibition of mammalian target of rapamycin complex 1 (mTORC1), both of which synergistically enhance fatty acid oxidation (FAO), mitochondrial biogenesis, and lipid clearance.
Inflammatory Response: NAFLD activates systemic inflammation through pro-inflammatory factors (tumor necrosis factor-alpha [TNF-α], IL-6, CRP), damaging vascular endothelia and promoting AS plaque formation.28–31 Direct anti-inflammatory actions of LKB1/AMPK include suppression of NF-κB signaling to reduce IL-6 levels,25,32 promotion of macrophage M2 polarization, 25 and autophagy induction via the AMPK/mTOR pathway.26,33 Secondary/synergistic effects involve SIRT1-mediated p65 deacetylation, Nrf2 activation (driving antioxidant gene transcription), and further mTORC1 suppression to limit inflammatory gene expression. AMPK activators also reduce HDL inflammatory index and myeloperoxidase/paraoxonase-1 ratios, 25 systemically inhibiting inflammatory damage.
Endothelial Dysfunction: NAFLD induces endothelial injury via oxidative stress and lipid peroxidation, exacerbated by IR and elevated free fatty acids.28,30 Direct AMPK effects include inhibition of protein kinase C (PKC) signaling to reduce ox-LDL-induced endothelial apoptosis 33 and enhancement of eNOS phosphorylation for vasodilation. 32 Secondary effects include SIRT1 activation (independently improving endothelial mitochondrial function) and Nrf2-driven reactive oxygen species (ROS) detoxification, which synergize with AMPK-mediated vascular protection.
Oxidative Stress: NAFLD increases hepatic oxidative stress, with malondialdehyde accumulation damaging hepatocytes and endothelia, promoting foam cell formation. Direct AMPK effects include suppression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity (via thioredoxin; uncoupling protein 2) and clearance of damaged mitochondria through ULK1-mediated autophagy. 33 Secondary contributions arise from Nrf2 activation (which induces antioxidant genes such as heme oxygenase-1, HO-1; NAD(P)H quinone dehydrogenase 1) and SIRT1 signaling, both of which can also be triggered by AMPK to amplify antioxidant defense.
Energy Metabolism Dysregulation: IR, a core mechanism linking NAFLD and AS, drives hepatic lipolysis and free fatty acids release, exacerbating dyslipidemia and endothelial injury. Direct AMPK actions include improving insulin sensitivity, enhancing glucose transporter type 4 (GLUT4) translocation, and suppressing hepatic gluconeogenesis. Secondary and independent pathways include peroxisome proliferator-activated receptor alpha/gamma (PPARα/γ) regulation of lipid oxidation, SIRT6 inhibition of NF-κB inflammation, and angiotensin-converting enzyme 2/angiotensin-(1-7) axis modulation of renin–angiotensin system activity. 34 Exosome-mediated organ crosstalk exacerbates metabolic disorders: hepatocyte-derived exosomes impair cholesterol efflux, 19 while AS plaque-derived exosomes worsen hepatic lipid deposition and inflammation.24,35 Immune cells further disrupt liver–vascular crosstalk via exosomal cytokines or non-coding RNAs.18,35 Thus, both primary LKB1/AMPK signaling and secondary metabolic–immune pathways jointly underlie NAFLD–AS comorbidity (Figure 1).

Hepato-Vascular Pathological Interplay in NAFLD-AS Comorbidity. Annotations: LCFA: Long Chain Fatty Acid; CHOL: Cholesterol; WAT: White Adipose Tissue; PAF: Platelet Activating Factor; LM: Leukocyte Migration; ICI: Inflammatory Cell Infiltration;Endo MT: Endothelial Mesenchymal Transition; DM: Dysfunctional Mitochondria; MFI: Mitochondrial Fission Imbalance; IR: Insulin Resistance. Created with BioRender.com.
Biological Functions of the LKB1/AMPK Signaling Pathway
Basic Overview of the LKB1/AMPK Pathway
The LKB1/AMPK signaling pathway is a well-established regulatory hub in metabolic and inflammatory diseases, with extensive evidence from cellular, animal, and human studies linking its dysfunction to NAFLD, cardiometabolic disorders, and cancer.36,37 AMPK, a heterotrimeric protein kinase (composed of α, β, and γ subunits, forming 12 isoforms), maintains energy homeostasis by sensing intracellular ATP/AMP ratios. LKB1, a key upstream kinase, directly activates AMPK by phosphorylating Thr172 (Thr174 in α2). 37 Direct regulation occurs through AMP binding, conformational activation, and prevention of Thr172 dephosphorylation. Secondary regulation involves associated pathways such as SIRT1 (nicotinamide adenine dinucleotide-dependent deacetylase), mTOR (nutrient-sensing kinase), and Nrf2 (antioxidant transcription factor), which can be either AMPK-dependent or independent.
Role of LKB1/AMPK in Metabolic Regulation:
Lipid Metabolism
Direct mechanisms: The LKB1/AMPK axis directly phosphorylates ACC1/ACC2 at Ser79/Ser212, 36 38–41 reducing malonyl-CoA levels and thereby blocking fatty acid and cholesterol synthesis via HMG-CoA reductase (HMGR) inhibition, 42 while relieving malonyl-CoA-mediated suppression of CPT1 to promote mitochondrial long-chain fatty acid β-oxidation.36,38,43 AMPK also suppresses lipogenic transcription factors (SREBP1c; ChREBP) and downregulates fatty acid synthase (FASN) and Hmgcr expression, 39 44–46 and activates CPT1a to enhance fatty acid mitochondrial transport. 47
Secondary/Synergistic mechanisms: AMPK synergizes with the SIRT1–PGC-1α–PPARα axis (via SIRT1-mediated PGC-1α deacetylation) to upregulate CPT1a and acyl-CoA oxidase 1, promoting mitochondrial and peroxisomal β-oxidation.48–50 It interacts with mTORC1 inhibition (via tuberous sclerosis complex 2 (TSC2)/regulatory-associated protein of mTOR phosphorylation)51,52 and enhances nicotinamide adenine dinucleotide metabolism to boost oxidative capacity. 50 In the hypothalamus, LKB1/AMPK–pro-opiomelanocortin (POMC) neuron activation promotes white adipose tissue (WAT) browning via PR domain containing 16–PPARγ and suppresses hepatic SREBP1c.53,54 These secondary mechanisms can act independently of AMPK but usually enhance its lipid-lowering effects, contributing to immune regulation (eg, T-cell FAO support 47 and organ protection (eg, reversing hepatic steatosis and renal fibrosis.49,55
Inflammatory Response
Direct mechanisms: AMPK inhibits mTORC1 via TSC2 phosphorylation,36,56 blocks NF-κB and c-Jun N-terminal kinase (JNK) activation, and phosphorylates inhibitor of κB kinase α/β (IKKα/β) to prevent NF-κB nuclear translocation. It also reduces ROS production via PKC–NADPH oxidase inhibition and phosphorylates forkhead box protein O1/3 to induce superoxide dismutase 2 (SOD2)/catalase (CAT) expression.57,58
Secondary/Synergistic mechanisms: SIRT1-mediated p65 deacetylation and PGC-1α competition with p65 further suppress pro-inflammatory cytokines (TNF-α, IL-1β, IL-6).54,59,60 LKB1 maintains tricarboxylic acid (TCA) cycle homeostasis to control 2-hydroxyglutarate levels, modulating chromatin accessibility, while AMPK limits inflammatory transcription via poly (ADP-ribose) polymerase 1/histone deacetylase 5 inhibition.61,62 Nrf2 activation enhances antioxidant gene expression. 57 Tissue-specific effects include hypothalamic LKB1/AMPK stabilization of inhibitor of κBα to suppress transforming growth factor-beta1 (TGF-β1)–Toll-like receptor 4 (TLR4) signaling 53 and intestinal LKB1 maintenance of barrier integrity. 44 These secondary mechanisms act independently but synergize with AMPK to inhibitNLR family pyrin domain containing 3 (NLRP3) inflammasome activation, 58 modulate T helper 17/regulatory T (Th17/Treg) balance,61,62 and reduce lipotoxic inflammation.58,63
Endothelial Function
Direct mechanisms: AMPK phosphorylates eNOS at Ser1177 to enhance NO production,41,56 increases eNOS expression, and prevents uncoupling. It also suppresses pro-apoptotic JNK/p38 mitogen-activated protein kinase (MAPK) signaling and inhibits PKC–NADPH oxidase to reduce ROS.57,59
Secondary/Synergistic mechanisms: SIRT1 activation improves mitochondrial function and supports eNOS activity. 59 Nrf2-mediated oxidative stress control reduces endothelial inflammation. 57 LKB1/AMPK also downregulates adhesion molecules (vascular cell adhesion molecule 1, VCAM-1; intercellular adhesion molecule 1, ICAM1; E-selectin) and chemokines (monocyte chemoattractant protein 1, MCP-1; regulated on activation, normal T cell expressed and secreted), 59 and works with PPARα to maintain endothelial integrity—loss of this axis (eg, high-fat diet) impairs function, whereas PPARα agonists restore it. 52
Mitochondrial Function
Direct mechanisms: LKB1/AMPK activates PGC-1α (with nuclear respiratory factor 1) to drive mitochondrial biogenesis, improves oxidative phosphorylation, FAO, and respiratory capacity.52,63 It phosphorylates ULK1 for mitophagy and mitochondrial fission factor (MFF) for mitochondrial fission, maintaining network dynamics.36,51,64
Secondary/Synergistic mechanisms: SIRT1 deacetylates PGC-1α to couple mitochondrial biogenesis with quality control, 51 and Nrf2 induction boosts mitochondrial antioxidant defenses.52,65 During mitochondrial dysfunction, LKB1 restores TCA activity via NADH metabolism and phosphoglycerate dehydrogenase regulation. LKB1/AMPK deficiency in type 2 innate lymphoid cells causes oxidative phosphorylation impairment and ROS accumulation, suppressing IL-5/IL-13 via B-cell lymphoma-extra large (Bcl-xL) inhibition and ROS–nuclear factor of activated T-cells–programmed cell death protein 1 (PD-1) activation, which can be rescued by PD-1 blockade.61,66
Energy Metabolism
Direct mechanisms: AMPK improves insulin sensitivity by enhancing β-cell insulin synthesis/secretion, upregulating GLUT4 translocation, and suppressing hepatic glucose-6-phosphatase (G6Pase)/phosphoenolpyruvate carboxykinase (PEPCK).36,38,39,56 It modulates glycolysis (6-phosphofructo-2-kinase activation during ischemia, Warburg effect suppression in tumors) and reduces energy expenditure on protein synthesis.39,42,56
Secondary/Synergistic mechanisms: peroxisome proliferator-activated receptors, SIRT1, and SIRT6 modulate energy metabolism in cooperation with AMPK. 50 LKB1 regulates hypothalamic POMC neurons to control appetite and glucose balance,53,55 and stabilizes PD-1/glucocorticoid-induced TNFR-related protein in Treg cells to modulate immune metabolism.32,55 The Nrf2 axis enhances antioxidant defenses (SOD2/CAT).41,54,63 These pathways form a metabolic–inflammation–immune network that can function independently but collectively sustain systemic energy homeostasis.
The integrated multi-organ regulatory roles of LKB1/AMPK signaling in NAFLD and AS are illustrated in Figure 2. The following section explores the therapeutic potential of various natural products that target this multifaceted LKB1/AMPK signaling hub, highlighting their unique mechanistic features and clinical applications in NAFLD and AS.

Mechanistic Network of LKB1/AMPK-Mediated Multi-Organ Regulation in NAFLD and AS. Annotations: ACC: Acetyl-CoA Carboxylase; SREBP1c: Sterol Regulatory Element-Binding Protein 1c; CPT1A: Carnitine Palmitoyltransferase 1A; PGC-1α: Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha; PPARα: Peroxisome Proliferator-Activated Receptor Alpha; LCFA: Long Chain Fatty Acid; CHOL: Cholesterol; WAT: White Adipose Tissue; BAT: Brown Adipose Tissue; TSC2: Tuberous Sclerosis complex 2; IKKα/β: Inhibitor of Nuclear Factor Kappa-B Kinase Subunit Alpha/beta; mTORC1: Mechanistic Target of Rapamycin complex 1; NF-κB: Nuclear Factor Kappa B; TNF-α: Tumor Necrosis Factor-Alpha; IL-1β: Interleukin-1 beta; IL-6: Interleukin-6; IL-17: Interleukin-17; VEGF: Vascular Endothelial Growth Factor; eNOS: Endothelial Nitric Oxide Synthase; VCAM-1: Vascular Cell Adhesion Molecule 1; ICAM-1: Intercellular Adhesion Molecule 1; PAF: Platelet Activating Factor; LM: Leukocyte Migration; ICI: Inflammatory Cell Infiltration;Endo MT: Endothelial Mesenchymal Transition; ULK1: Unc-51 Like Autophagy Activating Kinase 1; MFF: Mitochondrial Fission Factor; Bcl-xL: B-Cell Lymphoma-Extra Large; mtDNA: Mitochondrial DNA; DM: Dysfunctional Mitochondria; MFB: Mitochondrial Fission Balance; ILC2 mt: Type 2 Innate Lymphoid Cells Mitochondrial; NPY: Neuropeptide Y; AgRP: Agouti-Related Peptide; POMC: Pro-Opiomelanocortin; CART: Cocaine- and Amphetamine-Regulated Transcript; FoxO1: Forkhead box Protein O1; GLUT4: Glucose Transporter Type 4; G6Pase: Glucose-6-Phosphatase; PEPCK: Phosphoenolpyruvate Carboxykinase. Created with BioRender.com.
Therapeutic Potential of Natural Products Targeting LKB1/AMPK Pathway in NAFLD and AS
LKB1/AMPK pathway serves as a central signaling hub, integrating regulation of lipid metabolism, inflammation, oxidative stress, endothelial function, and mitochondrial homeostasis, as detailed in Section 3. The therapeutic potential of the natural products discussed below primarily arises from their ability to activate this master regulatory axis, thereby invoking its broad spectrum of beneficial downstream effects. Rather than reiterating these common effects (eg, ACC phosphorylation, SREBP1c suppression, eNOS activation), this section will highlight the unique mechanisms, direct molecular targets, clinical translation evidence, and specific bioavailability challenges associated with each compound or herbal extract. The overarching goal is to showcase their multi-targeted and synergistic approaches to disrupting the NAFLD-AS comorbidity network.
Multi-Target Effects of Single Herbal Medicines
Rhizome-Derived Herbs
Turmeric-derived bioactive compounds (tetrahydrocurcumin [THC], fermented turmeric, curcumin) exert their effects primarily by directly activating the LKB1/AMPK axis.67–71 Unique Mechanisms and Synergies: A unique and direct plaque-stabilizing mechanism of curcumin is the inhibition of macrophage extracellular matrix metalloproteinase inducer (EMMPRIN)/matrix metalloproteinase (MMP) expression, which is independent of its AMPK-mediated anti-inflammatory effects. 71 THC further demonstrates a strong secondary benefit by improving insulin sensitivity through insulin receptor substrate–phosphoinositide 3-kinase–protein kinase B (IRS–PI3K–Akt) signaling. 67 Clinical Evidence and Challenges: Despite promising preclinical data, clinical evidence is mixed. A randomized controlled trial (RCT) found 1500 mg/day curcumin was not superior to lifestyle intervention alone for NAFLD inflammation, 72 while meta-analyses suggest benefits for metabolic parameters. 73 This discrepancy underscores the critical challenge of its low oral bioavailability, driving the development of novel formulations like BCM-95® 72 and nanoparticles.74–76
Ginseng-derived bioactive compounds (ginsenosides Rg5, Rb2, compound K [CK], Rg1, Rb1, Rg3) mediate their effects through direct activation of the upstream kinase LKB1.77–82 Unique Mechanisms and Synergies: Rg3 promotes macrophage M2 polarization through PPARγ activation, showcasing an immunomodulatory mechanism that complements its AMPK-mediated effects.79,81 Rb1 enhances endothelial function via the SIRT1/AMPK axis, providing a vasoprotective synergy. 82 Clinical Evidence and Challenges: Clinical trials for pure ginsenosides are scarce. A major translational bottleneck is the extremely low bioavailability (<1%) of highly active metabolites like ginsenoside CK,83–85 necessitating strategies like albumin nanoparticles. 86
Salvia miltiorrhiza-derived bioactive compounds (salvianolic acid A [SAA], sodium tanshinone IIA sulfonate [STS], Radix Rubiae Paeoniae [RRP], puerarin [PA]) exert multi-target effects via the LKB1/AMPK axis.87–91 Unique Mechanisms and Synergies: SAA uniquely inhibits ferroptosis, linking AMPK activation to a novel cell death pathway in NAFLD. 87 STS activates the AMPK/Nrf2/HO-1 pathway, providing a potent antioxidant effect. 89 Clinical Evidence and Challenges: Robust clinical trial data for NAFLD are lacking. 92 A significant hurdle is the poor oral bioavailability of its key components, 92 spurring the development of innovative co-delivery systems like metal-phenolic networks93,94
Leaf-Derived Herbs
Artemisia annua extracts (scopoletin [SCO]) and artemisinin function through the LKB1/AMPK pathway.95–97 Unique Mechanisms and Synergies: Artemisinin demonstrates a compelling multi-faceted immunomodulatory role by suppressing the NLRP3 inflammasome through the AMPK–NF-κB–NLRP3 pathway. 97 Clinical Evidence and Challenges: Clinical trials for NAFLD/AS are very limited. 98 Repurposing this antimalarial drug faces challenges: low bioavailability (19-35%), individual metabolic variations, and potential toxicity at high doses. 98
Ginkgolide C (GC) and Ginkgo biloba extract (GbE) act via LKB1/AMPK.99,100 Unique Mechanisms and Synergies: GC additionally enhances mitochondrial function through the LKB1/AMPK–PGC-1α axis. 99 GbE's effects are further mediated via Akt/eNOS signaling and suppression of p38 mitogen-activated protein kinase (MAPK)/NF-κB. 100
Flower-Derived Herbs
Saffron-derived bioactive compounds (crocin, crocetin, safflower yellow A [SYA]) show efficacy via LKB1/AMPK. 90 101–103 Unique Mechanisms and Synergies: Crocetin engages the SIRT1/AMPK pathway to improve lipid profiles. 103 It also suppresses vascular inflammation by inhibiting NF-κB activation.102,103 Clinical Evidence and Challenges: Clinical studies, particularly in coronary artery disease patients, show promise. Crocetin (10 mg/day) improved lipid profiles and homocysteine, 103 while crocin (30 mg/day) modulated gene expression (SIRT1/AMPK↑; lectin-type oxidized LDL receptor 1/NF-κB↓) and reduced ox-LDL. 104 Saffron is generally safe, but its dose optimization is complex due to the different potencies of its compounds.103,105
These findings collectively indicate that natural products exert protective actions against NAFLD and AS by simultaneously modulating multiple targets and coordinating cross-organ crosstalk through the LKB1/AMPK pathway. A schematic overview of these multi-organ and multi-target mechanisms is illustrated in Figure 3.

Multi-Target and multi-Organ Protective Mechanisms of Natural Products Against NAFLD and AS via the LKB1/AMPK Pathway. Annotations: SIRT1: Sirtuin 1; PGC-1α: Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha; mTOR: Mechanistic Target of Rapamycin; ACC: Acetyl-CoA Carboxylase; CPT1A: Carnitine Palmitoyltransferase 1A; SREBP1c: Sterol Regulatory Element-Binding Protein 1c; Hmgcs2: 3-Hydroxy-3-Methylglutaryl-CoA Synthase 2; HMGR: 3-Hydroxy-3-Methylglutaryl-CoA Reductase; eNOS: Endothelial Nitric Oxide Synthase; NF-κB: Nuclear Factor Kappa B; IL-6: Interleukin-6; TNF-α: Tumor Necrosis Factor-Alpha; ULK1: unc-51 Like Autophagy Activating Kinase 1; HO-1: Heme Oxygenase 1; PPARα: Peroxisome Proliferator-Activated Receptor Alpha; PPARγ: Peroxisome Proliferator-Activated Receptor Gamma; IRS: Insulin Receptor Substrate; FASN: Fatty Acid Synthase; SCD1: Stearoyl-CoA Desaturase-1; DGAT1: Diacylglycerol O-Acyltransferase 1; AP-1: Activator Protein 1; p53: Tumor Protein p53; p21: Cyclin-Dependent Kinase Inhibitor 1A; IGFBP1: Insulin-Like Growth Factor Binding Protein 1; TGFβ: Transforming Growth Factor beta; CTGF: Connective Tissue Growth Factor; Col1a1: Collagen Type I Alpha 1 Chain; NLRP3: NLR Family Pyrin Domain Containing 3; ICAM-1: Intercellular Adhesion Molecule 1; VCAM-1: Vascular Cell Adhesion Molecule 1; LXRα: Liver X Receptor Alpha; M1→M2 Polarization: Macrophage M1 to M2 Polarization; THC: Tetrahydrocurcumin; GC: Ginkgolide C; GbE: Ginkgo biloba Extract; SYA: Safflower Yellow A; SAA: Salvianolic Acid A; STS: Sodium Tanshinone IIA Sulfonate; RRP: Radix Rubiae Paeoniae; PA: Puerarin; SCO: Scopoletin. Created with BioRender.com.
Targeted Regulation of Monomeric Compounds
Polyphenolic Compound
Luteolin ameliorates pathology through the nicotinamide phosphoribosyltransferase (NAMPT)-Sirt1/AMPK axis.106–108 Unique Mechanisms and Synergies: Its effects are significantly reinforced by its unique action of upregulating NAMPT–Sirt1 signaling, leading to reduced forkhead box protein O1 (FOXO1) acetylation and improved lipid metabolism.106–108 Clinical Evidence and Challenges: Direct clinical evidence in NAFLD/AS is very limited. 109 A major challenge is that dietary intake is much lower than efficacious preclinical doses. 109
Baicalin (BAI) multi-dimensionally ameliorates pathology by targeting the LKB1/AMPK axis.110–112 Unique Mechanisms and Synergies: BAI activates the Nrf2/HO-1 antioxidant pathway and inhibits NF-κB-mediated inflammation.110,111 It also enhances insulin sensitivity via the AMPK–Akt/glycogen synthase kinase-3 beta (GSK-3β) axis. 112 Clinical Evidence and Challenges: There is a notable lack of clinical trials investigating baicalin for NAFLD or AS. 113 Its clinical translation is primarily hampered by low oral bioavailability. 113
Quercetin (Que) ameliorates pathology via the LKB1/AMPK axis. 97 114–116 Unique Mechanisms and Synergies: Que alleviates endoplasmic reticulum (ER) stress through the AMPK/protein phosphatase 2A (PP2A) axis. 115 It also promotes macrophage M2 polarization and inhibits the M1 phenotype via Akt signaling.114,116 Clinical Evidence and Challenges: An RCT in NAFLD patients showed that 500 mg/day quercetin for 12 weeks significantly reduced hepatic fat content (∼17.4%). 117 Its low and variable bioavailability presents a significant challenge.117,118
Xanthohumol (Xn) exerts therapeutic effects on NAFLD and AS by activating AMPK phosphorylation.119,120 Unique Mechanisms and Synergies: Xn induces Nrf2 nuclear translocation, enhances antioxidant enzyme activity, and inhibits NF-κB-driven cytokine release. 119 It further reduces MCP-1 expression. 120
Nobiletin (NOB) synergistically regulates lipid metabolism and inflammation via AMPK signaling.121,122 Unique Mechanisms and Synergies: NOB activates IRS-1/AKT and SIRT1-related pathways, thereby enhancing glucose uptake and modulating inflammatory responses. 121
EGCG exerts cross-disease therapeutic effects by activating LKB1/AMPK signaling.123–125 Unique Mechanisms and Synergies: EGCG restores fibroblast growth factor 21 (FGF21) signaling sensitivity and activates the adiponectin receptor 2 (AdipoR2)/SIRT1 axis.123,124 It also enhances antioxidative defenses via the Nrf2 pathway. 124 Clinical Evidence and Challenges: Clinical evidence for EGCG in NAFLD is limited and not yet conclusive. 126 Concerns about potential hepatotoxicity at high doses and its low bioavailability complicate dose optimization.126,127
Resveratrol (RSV) exerts multi-target therapeutic effects by activating the SIRT1-dependent LKB1/AMPK axis.128–135 Unique Mechanisms and Synergies: RSV promotes cholesterol efflux through the liver X receptor alpha (LXRα)/ABC-A1 axis.129,130 It inhibits inflammation via NF-κB/TLR4 suppression and upregulates antioxidative defenses.131–133 Clinical Evidence and Challenges: Clinical trial results are mixed. A meta-analysis found no significant effect on most NAFLD parameters, 136 while other RCTs show benefits.137,138 Its very low oral bioavailability is a major limitation.137,138
Gallic acid (GA) exerts multi-target therapeutic effects by activating the LKB1-dependent AMPK pathway.139–141 Unique Mechanisms and Synergies: GA downregulates lipid uptake genes and enhances antioxidant defenses via the Nrf2/HO-1 axis.139,140 In AS, GA improves endothelial function via AMPK–eNOS activation.140,141 Clinical Evidence and Challenges: Clinical data for gallic acid in NAFLD or AS is currently lacking. 142 Its development is challenged by high water solubility leading to low bioavailability. 142
Terpenoid Compound
Betulinic acid (BA) ameliorates NAFLD and AS by modulating the calcium/calmodulin-dependent protein kinase kinase–AMPK signaling axis.143–145 Unique Mechanisms and Synergies: BA suppresses mTOR–ribosomal protein S6 kinase signaling and activates PGC-1α-associated thermogenesis.143,144
Gastrodin exerts cross-disease regulatory effects through the LKB1/AMPK signaling axis.146–148 Unique Mechanisms and Synergies: Gastrodin activates the Nrf2/HO-1 antioxidant pathway and inhibits NF-κB-mediated inflammation.146–148 It also intervenes by inhibiting TGF-β/Smad2-driven fibrosis in NAFLD. 147
Alkaloid Compound
Berberine (BBR) is a premier example of a natural product with cross-disease efficacy through the LKB1/AMPK signaling axis.149–155 Unique Mechanisms and Synergies: Uniquely, a significant part of BBR's metabolic benefit originates from its ability to modulate gut microbiota and bile acid metabolism, a systemic effect that indirectly reinforces AMPK activation.104,106 Clinical Evidence and Challenges: Berberine is standout for its strong clinical validation. Multiple RCTs confirm its efficacy in improving liver enzymes, lipid profiles, and IR in NAFLD.156,157 The primary challenge remains its low bioavailability, motivating research into novel delivery systems and combination therapies.156,158,159
Glycoside Compound
Salidroside (SAL) exerts pleiotropic therapeutic effects by targeting the LKB1/AMPK signaling axis.160–162 Unique Mechanisms and Synergies: SAL additionally regulates oxidative stress and inflammation by modulating the NLRP3 inflammasome.160,161
Nucleoside Compound
Cordycepin ameliorates pathology through multi-target modulation of the LKB1/AMPK signaling axis.163,164 Unique Mechanisms and Synergies: Beyond direct AMPK activation, Cordycepin reduces tumor necrosis factor (TNF) signaling activity and inhibits TGF-β/Smad-driven fibrotic responses. 163
Phenolic Derivative
6-Gingerol exerts pleiotropic therapeutic effects through the LKB1/AMPK signaling axis.165–167 Unique Mechanisms and Synergies: 6-Gingerol reduces oxidative stress and inflammation. 165 In AS, it alleviates vascular inflammation via NF-κB pathway inhibition and suppresses macrophage pyroptosis.166,167
Representative structures of these compounds are shown in Figure 4, and their classifications and mechanisms of action are summarized in Table 1.
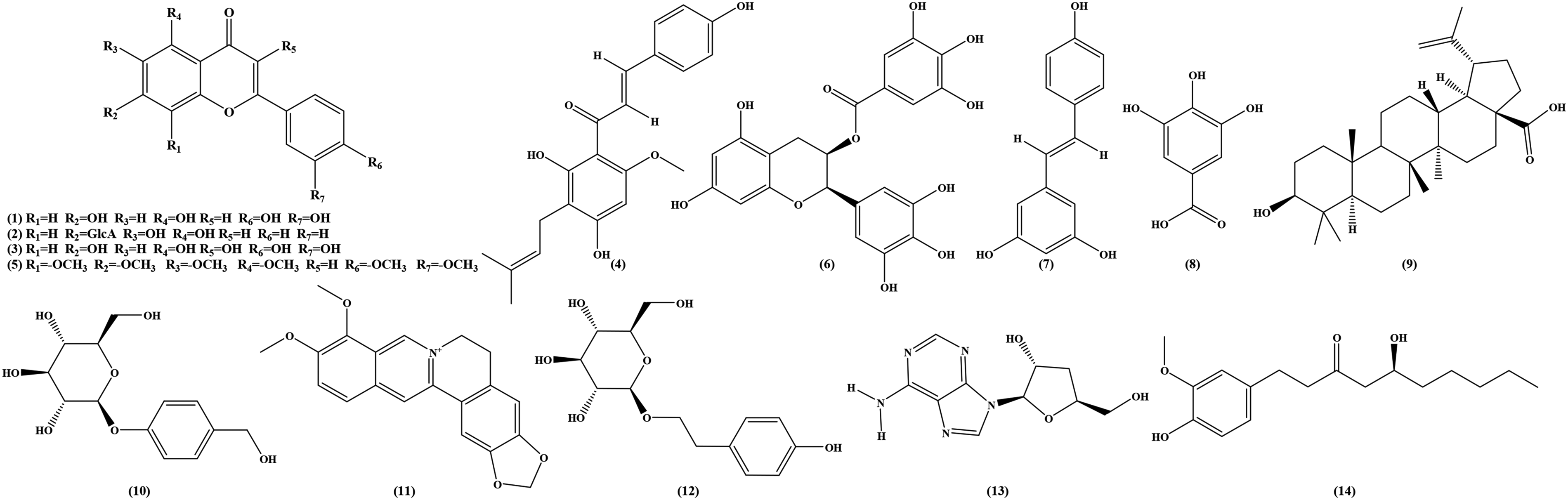
Bioactive Compounds Structures. (1) Luteolin; (2) Baicalin; (3) Quercetin; (4) Xanthohumol; (5) Nobiletin; (6) Epigallocatechin Gallate; (7) Resveratrol; (8) Gallic Acid; (9) Betulinic Acid; (10) Gastrodin; (11) Berberine; (12) Salidroside; (13)Cordycepin; (14) 6-Gingerol.
Comprehensive Summary of Natural Products Targeting the LKB1/AMPK Pathway and Associated Mechanisms in NAFLD and AS Therapy.

Pharmacological and Translational Challenges of Natural Products Targeting the LKB1/AMPK Pathway
Bioavailability Limitations
Despite promising efficacy in preclinical models, many natural products modulating the LKB1/AMPK pathway suffer from low oral bioavailability. Poor aqueous solubility, rapid phase II metabolism, and high polarity (eg, polyphenols, ginsenosides, phenolic acids) restrict systemic exposure.86,168,169 For example, curcumin shows negligible plasma levels after oral administration due to poor solubility and rapid metabolism,170,171 while many ginsenosides exhibit oral bioavailability often <1%–5%.172,173 These properties necessitate supraphysiological doses in animal studies and complicate dose selection for human trials.
Pharmacokinetics and Metabolism
The pharmacokinetic (PK) behavior of LKB1/AMPK-activating compounds is complex and variable. Absorption is strongly influenced by formulation and route of administration.169,174 Distribution is further affected by plasma protein binding and tissue affinity.175,176 Many compounds undergo rapid hepatic metabolism and conjugation, or are transformed by gut microbiota into secondary metabolites with distinct activity.177–179 Elimination pathways, including biliary and renal excretion, often shorten half-life (eg, artemisinin) and limit sustained exposure.180–182 In addition, drug–drug or nutrient–drug interactions (eg, with B vitamins or chemotherapeutics) can markedly alter systemic exposure.183–186
Species and Disease-State Variability
Interspecies differences in absorption, metabolizing enzymes, transporter expression, and microbiota composition lead to divergent pharmacokinetics and pharmacodynamics (PD) between rodents and humans.170,171 For instance, ginsenoside metabolite profiles and time to maximum concentration differ substantially between mice and rats.178,182 Moreover, nanoformulations that significantly increase maximum concentration/area under the curve in rodents often fail to achieve comparable exposure in humans. 187 Disease conditions such as NAFLD and AS further influence absorption and metabolism, complicating extrapolation of preclinical efficacy to patients.
Translation Gaps from Animal to Human Studies
Natural products such as resveratrol, EGCG, and xanthohumol robustly activate LKB1/AMPK in experimental models.86,170,175 However, clinical trials have yielded mixed or modest outcomes.188,189 Discrepancies often arise from insufficient systemic exposure in humans compared with animal studies, 190 differences in active metabolite formation, 191 and reliance on simplified animal models that fail to capture the heterogeneity of human NAFLD and AS. 192
Future Perspectives
To overcome these barriers, future studies should integrate rigorous PK/PD characterization, including metabolite profiling.175,188 Optimization of delivery systems and use of structural analogs are needed to enhance systemic exposure. 190 Physiologically based pharmacokinetic modeling can help bridge preclinical and clinical data. 191 Furthermore, early clinical trials should ensure that human exposure levels approximate those producing efficacy in animal models. 192 These strategies are essential to harness the therapeutic potential of natural products acting through the LKB1/AMPK pathway.
Discussion
LKB1/AMPK Signaling Hub: A Tripartite Regulatory Core Integrating Metabolism, Inflammation, and Mitochondria
LKB1/AMPK pathway acts as a “central processor” of energy metabolism, orchestrating multi-dimensional regulatory roles in the comorbidity of NAFLD and AS by integrating pathological networks between the hepatic and vascular systems. From a mechanistic perspective, AMPK bidirectionally regulates lipid metabolism by phosphorylating ACC (inhibiting lipid synthesis) and activating PPARα/CPT1a (promoting fatty acid β-oxidation), thereby breaking hepatic lipotoxicity cycles. Concurrently, it enhances macrophage cholesterol efflux via ABC-A1/ABC-G1 upregulation, suppressing AS plaque core formation.36,38,39 Tissue-specific regulation is also evident: hypothalamic LKB1 activates the AMPK-POMC neuron-sympathetic axis to induce white adipose browning and suppress hepatic SREBP1c expression,51,53,54 while intestinal LKB1 deficiency disrupts mitochondrial FAO, leading to lipid accumulation. 44
In addition to metabolic control, AMPK exerts potent anti-inflammatory effects through mTORC1/NF-κB inhibition and SIRT1-mediated p65 deacetylation, while reshaping the Th17/Treg immune balance.55,59,61 On the mitochondrial side, the LKB1/AMPK-PGC-1α network drives mitochondrial biogenesis and Unc-51 like autophagy activating kinase 1 (ULK1)-mediated mitophagy, while alleviating ROS-mediated vascular endothelial injury.43,52,63 Collectively, this “metabolism–inflammation–mitochondria” triad establishes a mechanistic foundation for therapies targeting LKB1/AMPK.
Liver–Vascular Axis Crosstalk: Exosomal miRNA-Mediated Communication
The vicious cycle of the liver–vascular axis is not solely dependent on classical circulating lipids or cytokines, but also mediated through exosomal microRNAs (miRNAs). Hepatocyte-derived small extracellular vesicle (sEV)-miR-30a-3p suppresses ABC-A1 expression, reducing macrophage cholesterol efflux and promoting coronary plaque formation. 35 Conversely, AS plaque-derived exosomes carrying receptor-interacting protein kinase 1 messenger RNA (mRNA) activate hepatic NLRP3 inflammasomes, accelerating NAFLD progression. 28 This bidirectional communication establishes a “hepatic lipotoxicity → vascular inflammation → hepatic fibrosis” feedback loop.
The LKB1/AMPK pathway may disrupt such pathological signaling by modulating exosomal cargo (eg, reducing miR-30a-3p secretion) and restoring mitochondrial function.35,61 Emerging interventions such as curcumin nanoparticles and RSV exemplify how natural compounds, acting through LKB1/AMPK activation, remodel inter-organ communication and immune microenvironments.67,71,107,193
Multi-Target Intervention by Natural Products: From Molecular Synergy to Clinical Translation Challenges
Natural products, with their multi-component and multi-target characteristics, hold unique promise in NAFLD-AS comorbidity. For example, ginsenoside Rg5 suppresses hepatic lipogenesis via the AMPK/mTOR axis while promoting FAO, STS activates both the AMPK/p53/p21 and Nrf2/HO-1 pathways, BBR exhibits liver-targeting with vascular benefits, and RSV simultaneously induces autophagy and improves endothelial function.69,70,72,79,106,107122–124,193
Despite these advances, clinical translation remains hindered by poor bioavailability, insufficient organ selectivity, and unresolved synergy among multiple components. New technologies provide potential solutions:
Nanocarriers improve solubility, bioavailability, and tissue targeting (eg, curcumin- or ginsenoside-loaded nanoparticles).170,171 Gut microbiota modulation leverages host–microbiome interactions to indirectly enhance AMPK activation.104,106 Structural modifications and bioenhancer combinations increase systemic exposure and therapeutic efficacy.179,191
Conclusion and Limitations
In conclusion, the LKB1/AMPK pathway integrates metabolic, inflammatory, and mitochondrial regulation, forming a central hub in the progression of NAFLD-AS comorbidity. Exosomal miRNA-mediated liver–vascular communication further amplifies disease burden, while natural products—through their multi-target and systemic regulatory potential—offer promising therapeutic opportunities.
However, several limitations must be acknowledged. Current evidence is largely derived from preclinical studies with heterogeneous models, and pharmacokinetic and pharmacodynamic profiles of many natural products remain insufficiently characterized. Moreover, the precise interactions among multi-component herbal preparations are still poorly understood.
Looking forward, emerging technologies such as nanocarrier-based delivery systems, gut microbiota modulation, and rational structural modification represent transformative strategies to overcome the long-standing barriers of bioavailability and organ specificity. By integrating these approaches, natural product–based interventions targeting the LKB1/AMPK axis may ultimately achieve successful clinical translation and provide novel therapeutic solutions for patients with NAFLD-AS comorbidity.
Footnotes
Abbreviations
Ethical Approval
Ethical Approval is not applicable for this article.
Author Contributions
Conceptualization, J.-P.Z. and J.-X.K.; writing—original draft preparation, J.-X.K.; writing—review and editing, J.-X.K.; visualization, T.W.; supervision, H.C.; project administration, J.-P.Z.; funding acquisition, J.-P.Z. All authors have read and agreed to the published version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Hunan Provincial Training Program for Young Core Teachers in Higher Education Institutions, University-level Scientific Research Project Funded by Hunan University of Chinese Medicine, Scientific Research Project Funded by Hunan Provincial Department of Education, (grant number 2024XJZC017, 23B0375).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data related to this study are presented in this publication.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.