
Abstract
Introduction
Diabetes mellitus is a worldwide health concern due to chronic glucose-related metabolic disorder. The previous evidences from clinical trials in the literature indicate that the disease not only results in hyperglycemia-related conditions but also might induce further complications.1–5 Based on the mechanism, it can be categorized into two main types: type 1 (by pancreatic β-cells deterioration) and type 2 (by insulin-metabolic abnormal activity); the latter accounts for 90–95% of cases. 6 Despite the well-rounded knowledge, the certain cause is still highly debated among the scientific community, possibly due to genetic abnormalities, pathologic disorders, clinical conditions, or gestational failures. 7 In effect, the condition is putting significant burden on the healthcare system.
Until now, diabetes mellitus has been treated by symptomatic relief rather than therapeutic cure. The current medicines available on the market only serve as the remedies. They only provide temporary effects against undesirable activities of certain biological enzymes, such as insulin- and glucose-based enzymes. For instance, the phosphorylation process is known to be catalyzed by protein tyrosine phosphatase 1B (PTP1B), which further stimulates the responsiveness of insulin in cells to uptake glucose. 8 On the other side, α-glucosidase is the bio-catalyst for breaking down -(1/4) and -(1/6) bonds in starch and disaccharide molecules through hydrolysis; meanwhile, α-amylase is the major form of salivary and pancreatic amylase found in humans and other mammals, responsible for the hydrolysis of α-linked polysaccharides (eg starch and glycogen) into shorter chains (eg dextrins and maltose). Their physicochemical roles induce the postprandial spikes from digestive carbohydrates.9,10 Given that, a solution to reduce glucose uptake can be possible; in particular, negative regulatory effects on the hyperglycemic activity or insulin signaling pathway are achievable with effective bio-inhibition against these proteins. In the commercial market, many hypoglycemic drugs have been introduced targeting glucosidases (eg Sulfonylureas, Biguanides, or Acarbose11,12) or the insulin-related enzyme (e.g Trodusquemine 13 ). However, they are known either with moderate-to-severe side effects, such as diarrhea and flatulence, or for its expensive price. Therefore, looking for alternative anti-diabetic agents, especially whose sources are ubiquitous in nature and friendly to the body, is still needed.
Goat's beard (Rau re in Vietnamese) has the scientific name of Aruncus dioicus (Walter) Fernald (synonyms A. sylvester Kostel. ex Maxim.; A. vulgaris (Maxim.) Raf. ex H. Hara), belonging to the family Rosaceae.14,15 This species is widely distributed in temperate regions of the Northern Hemisphere from Central Europe and the Caucasus to the Himalayas, China, Korea, Japan, Russia, Korea (Ulleung-do) and North America.16–20 It is a perennial herb up to 3 m tall with drooping flowers with numerous small, single, capsule, and sepalless clusters. 21 Goat's beard is known as a source of food, cosmetics, and medicinal plants in traditional Korean and Chinese medicine.22–28 However, regarding the Vietnamese A. dioicus, the knowledge on the active components and the biological activities is still poorly collected in the literature.
Evidence from preceding studies in the literature revealed the potential of the plant given its both compositional richness and bio-active diversity. Regarding the chemical bioavailability, a variety of phytochemicals have been identified in the aerial part and underground part of Goat's beard, including quercetin and kaempferol derivatives,29–33 3,4-dicaffeoylglucopyranosides,31,33 phenolic acids, especially hydroxycinnamic acid derivatives,33–36 tannins, 37 monoterpenoids29,32 and cyanide compounds (mainly prunasin).29,38 Particularly, the edible young shoots were found as a rich source of polyphenols; in which, 24 polyphenols were detected in the study by Fusani et al in 2016. 33 Further, Zhao et al reported the isolation of palmitic acid, 10-nonacosanol, pentacosan-1-ol, phytol, β-sitosterol, β-sitosterol-3-O- β-D-glucopyranoside, 2,4-dihydroxycinnamic acid, and hyperoside. 32 Some other works have demonstrated to present of lipophilic compounds, ie steroids, fatty acids, alcohols, 32 and caffeic acid derivatives. 33 In terms of bio-active potential, experimental findings from numerous studies indicate a profound diversification, including cytotoxic, anti-inflammatory, antioxidant, anti-cancer, antibacterial, neuroprotective, and anti-diabetic activities.20,23,28,29,39–42 Besides, the plant is rich in saponins, which are thought to be promising for the treatment of chronic degenerative diseases. The whole plant was used as the supplemental product for detoxification, prevent of tonsillitis, and stamina boost. 24 Several in vivo studies revealed the reduction of lipid accumulation and the improvement of insulin resistance, observed from C57BL/6 mice fed with high-fat diet supplemented with this plant.43,44 Also, it seemed to induced weight loss in goats 45 and alleviated streptozotocin-induced diabetes in rats. 41 Therefore, A. dioicus is considered as a potential natural source for anti-hyperglycemic candidates given its biochemical characteristics and evidences of anti-diabetic effects in particular.
However, the high chemical bioavailability and biological versatility, on the downside, might pose significant challenges in terms of experimental trials; therefore, cost-friendly and time-effective intervals, preliminarily examining the chemo-bio-pharmacological suitability of the extracted phytochemicals, are necessary. Given this, in silico research is of a propitious approach, which is able to harness the computer power to compensate for equivalent lab-based wet screening. In fact, the promising candidates can be quickly selected with desirable properties by different computational tools. For instance, based on molecular mechanics, docking technique with its simple computational algorithms can be an effective method for prediction of ligand-protein interaction; the cons often refer to its neglect of bio-medium factors, eg dipole compatibility or polar sensibility. Nevertheless, the former can be complemented by the results from quantum chemical calculations; while, the latter can be solved by the incorporation of proper arguments on physicochemical properties.46,47 Also, quantum chemical computation might provide other useful molecular properties for argument on intermolecular and chemical tendencies. Furthermore, pharmacokinetics and pharmacological properties of a chemical structure are able to be predicted by statistically regressive models, thus evaluating drug-likeness and medicinal chemistry friendliness. Altogether, these results can provide a reliable assessment of the biocompatibility and pharmaceutical suitability of numerous compounds in the cost-reduced and time-efficient manners.
In this work, A. dioicus was subject to a theory-experiment combinatory study. Firstly, lab-based experiments were carried out for its phytochemical characterization; the obtained structures were preliminarily used as the input for computational screening for biological and pharmacological potentials. Afterwards, a variety of computer-based platforms were utilized to predict the bio-chemo-pharmacological suitability and compatibility of the candidates; the arguments served as the justification for further in vitro tests. Finally, the phytochemicals were under accumulative isolation and subjected to enzymatic assays to evaluate their in-practice inhibitory activities against diabetes-related proteins, thus confirming the results from computational efforts. To better serve the readership, this report contents are structured by methodological contexts (ie experimental and computational parts) rather than following a chronological flow.
Results
Structural Elucidation
Compounds
Compound
Compound
Enzymatic Inhibition
Compounds (
in Vitro Inhibitory Activity of Compounds (
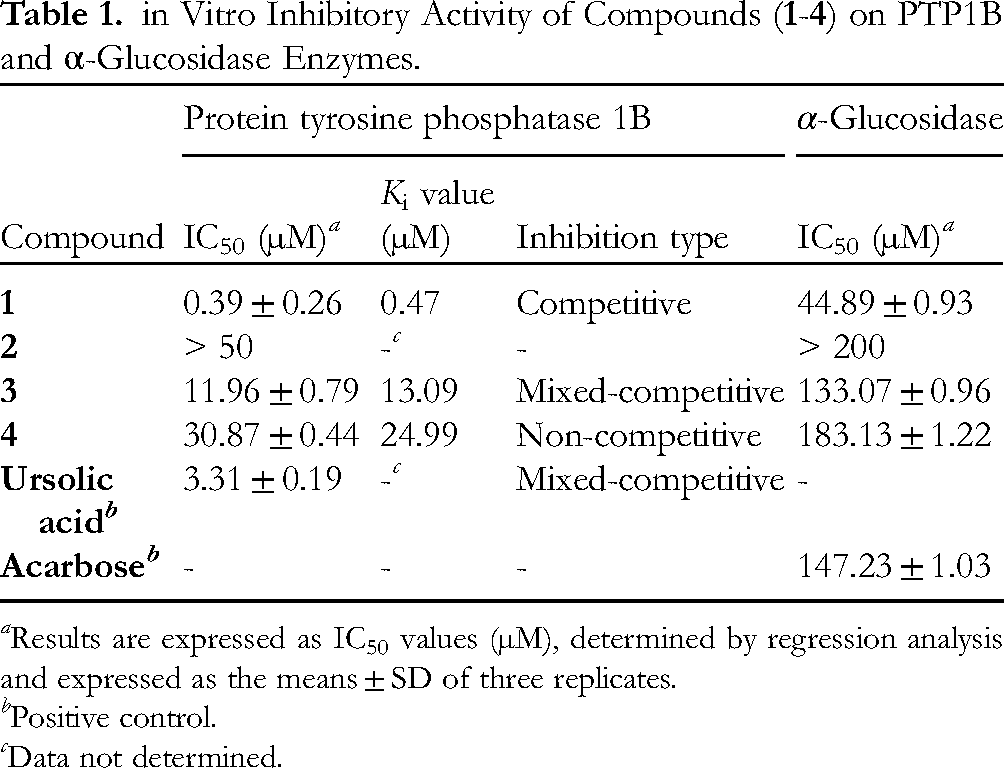
Results are expressed as IC50 values (µM), determined by regression analysis and expressed as the means ± SD of three replicates.
Positive control.
Data not determined.
Overall, all the tested compounds exhibited promising inhibition against PTP1B, except for
Figure 1 presents the analyses for Lineweaver-Burk and Dixon experiments under linear concentrations of substrate (p-NPP) with/without the presence of compounds
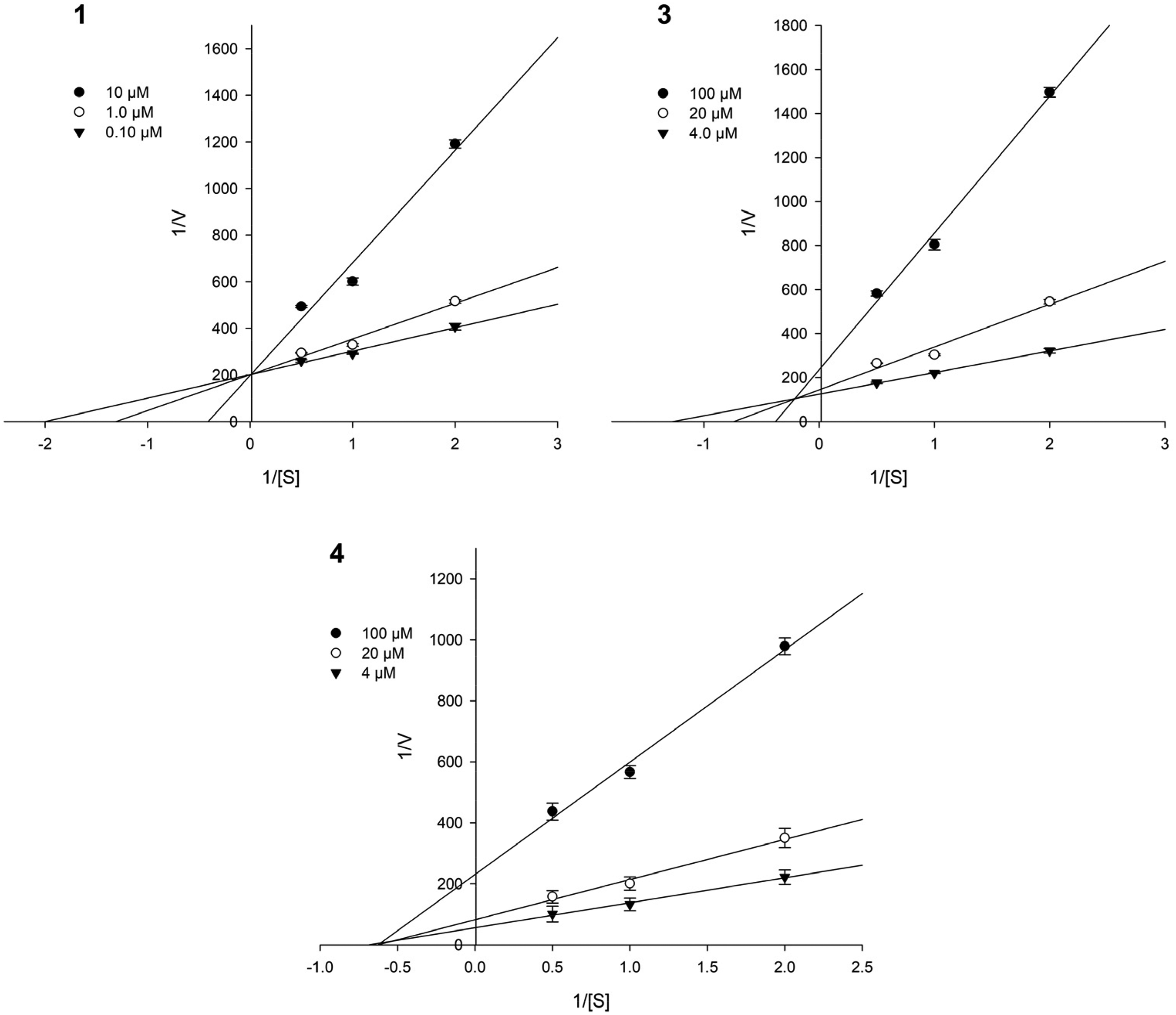
Lineweaver-Burk plots for the inhibition of compounds (
Figure 2 presents the analyses of Dixon plot experiments used to determine the Ki (inhibition constant) values from the x-axis where the straight lines intersected for the inhibitors.
56
In particular, the Ki values of

Dixon plots for active compounds (
Regarding α-glucosidase, the in vitro assays with the compounds (
Docking-Based Inhibitability
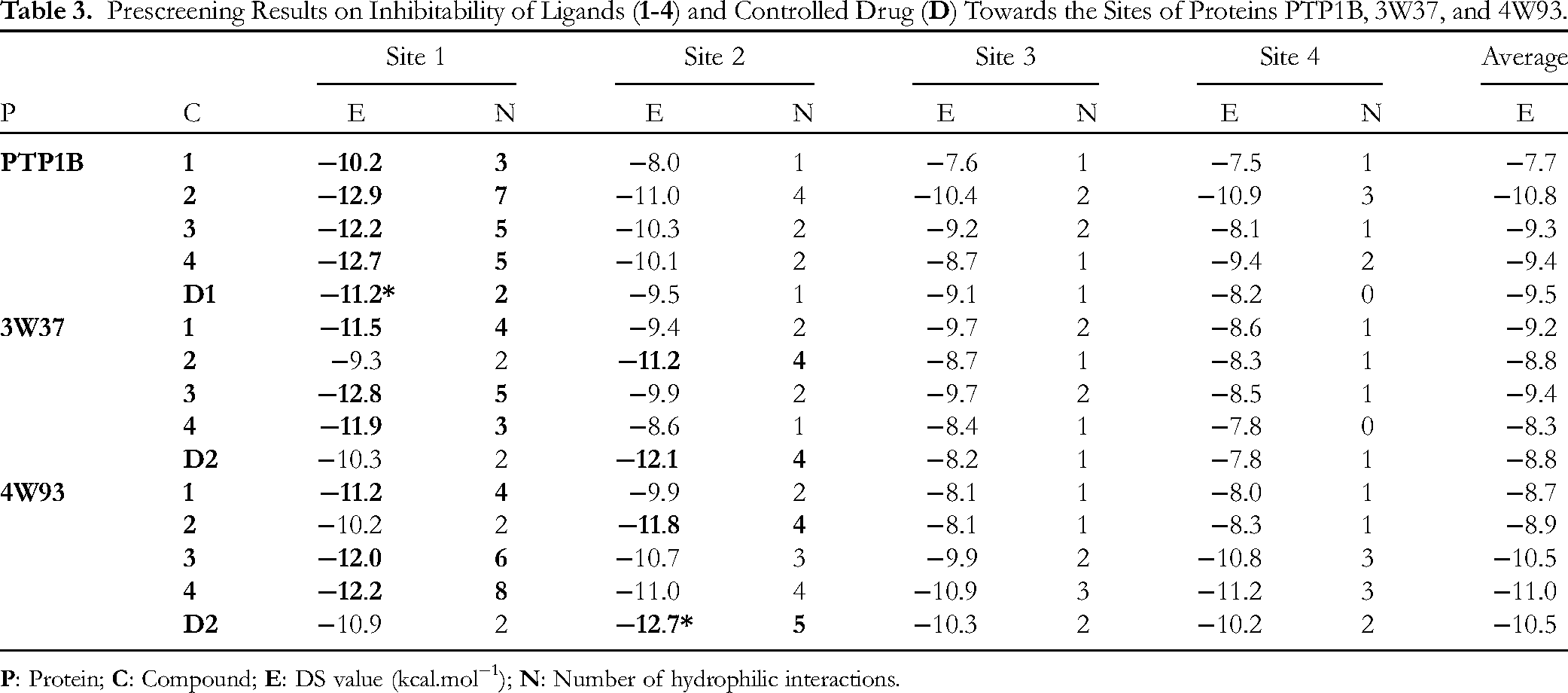
The results from docking simulation can be utilized for argument on the inhibitory effects of each compound (
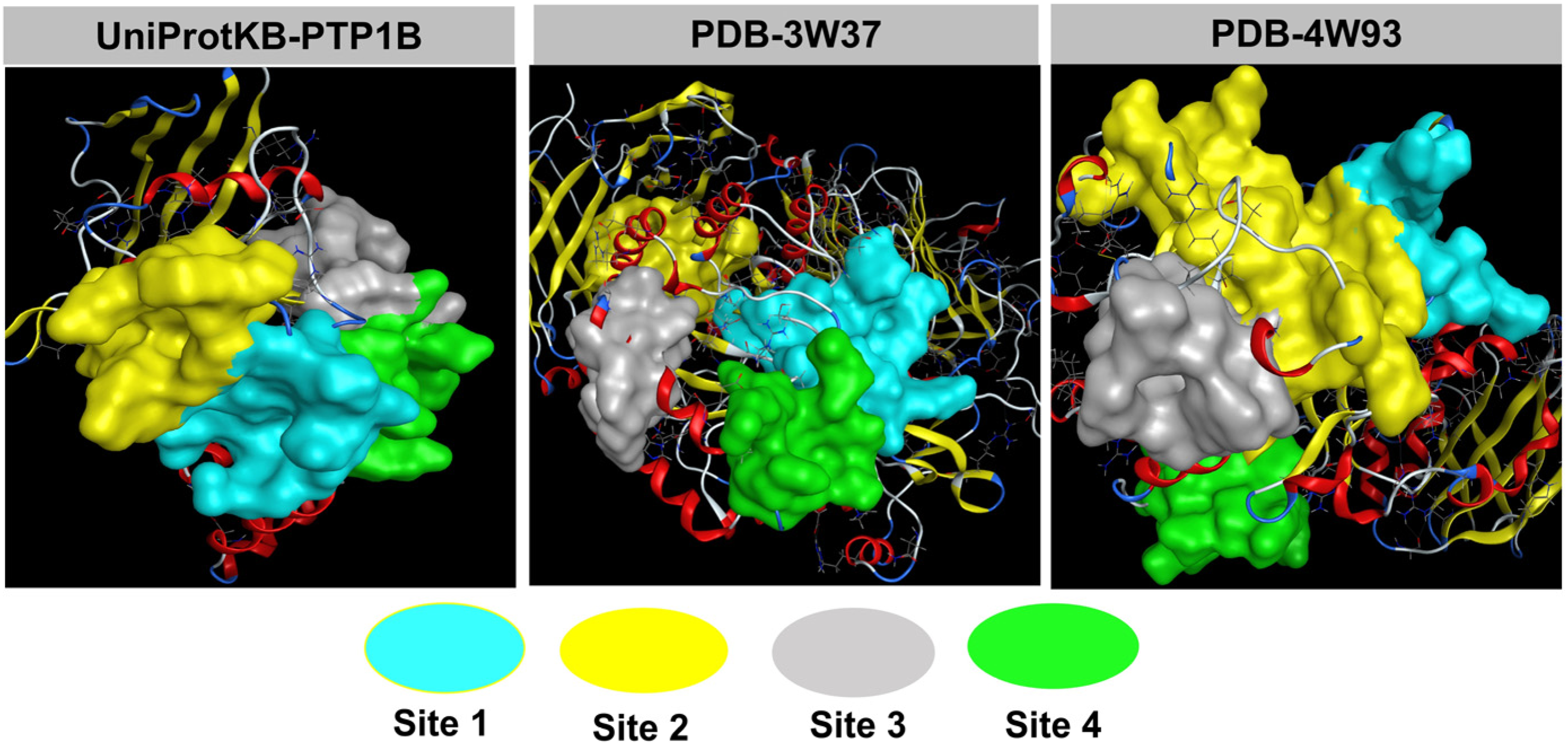
Quaternary structures of protein 3W37, PTB1B and 4W93 with the approachable sites by
The primary docking parameters are summarized in Table 2; the control drugs are ursolic acid (
Ground state electronic energy and dipole moment values of

The interacting configurations are also visually rendered, presented in Figure 4; these regard to the ligand-protein duo associated with highest DS value. The descriptive specification includes hydrogen-like bonding (dashed arrow), van de Waals interaction (blurry purple), and conformational fitness (dashed contour). The 3D configurations within the site indicate that the sites are relatively tight in relation to the size of the inhibitor. This means that significant structural modification on the current ligand structures are not recommended since the ligands would be oversized. Based on the 2D maps, it appears that the ligands exhibit favorable conformational fitness with the site features indicated by the continuousness of dashed contours. In-detail bonding parameters are summarized in Tables S1 (for ligand-PTP1B complexes), S2 (for ligand-3W37 complexes), and S3 (for ligand-4W93 complexes) in the supplemental information.

Visual presentation and in-pose interaction map of ligand-PTP1B, ligand-3W37, and ligand-4W93 inhibitory structures.
Quantum-Chemical Properties
The results from quantum calculation provide the preliminary view on the bio-medium compatibility and intermolecular interactability of the candidates based on ab initio insights of their chemical properties. Unlike docking, this argument concerns only the candidates (
The optimized geometries of the bioactive compounds are shown in Figure 5. In general, the input structures can be easily converged without encountering geometrical constraints or abnormal bonding angles and lengths. This is often of the characteristics of natural compounds; thereby in-turn also validating their spectroscopic characterisation and structural elucidation from the experimental characterization.

Geometrically optimised structures of
The main molecular properties, ie ground state energy and dipole moment, are summarized in Table 3. Firstly, the energy represents the overall chemical activeness of the host molecule; in the interpretation, the lower value equals to the less likeliness that the molecule might react with the body chemicals before reaching the targeted protein, thus more likely that it can retain the structural characteristics and biological properties. Given this argument,
Prescreening Results on Inhibitability of Ligands (
Molecular electronic potential (MEP) maps of the structures are given in Figure 6, providing the information on the distribution of electrostatic potential. By convention, reddish regions have negative tendency; bluish regions have positive tendency; greenish regions hold neither of the tendencies. From the conceptual view, the distribution of the chemical activities over each molecular plane can be argued for its flexibility when in interaction with external structures, which is especially useful in the scope of this study as protein-based sites often have arbitrary and unpredictable configurations. It can be seen that all the molecules (especially
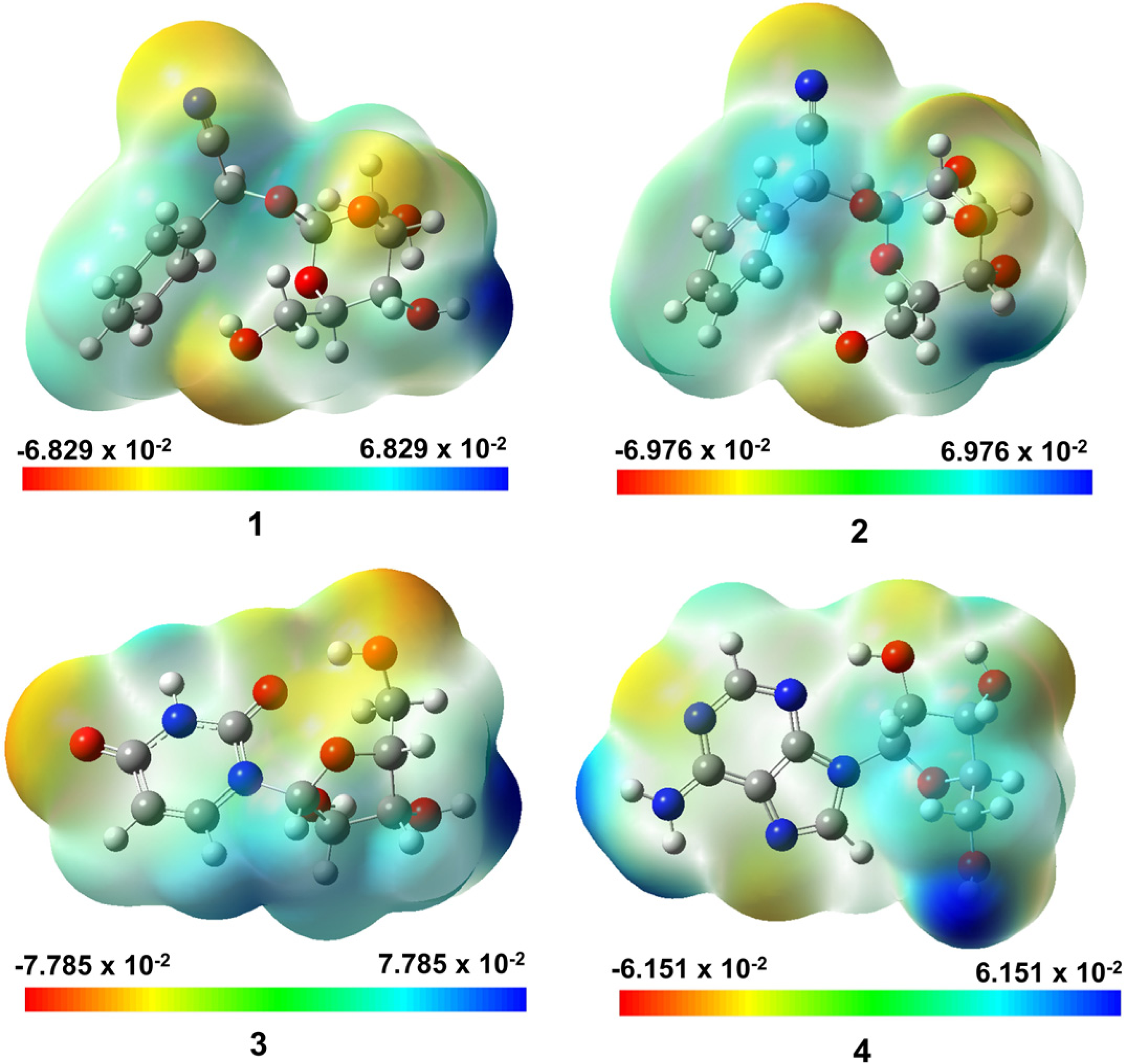
Molecular electrostatic potential (MEP) formed by mapping of total density over the electrostatic potential of
Physicochemical Properties
The physicochemical properties of the compounds are given in Table 4. QSARIS-derived physical properties include molecular mass (Da), polarizability (Å3), size (Å), and dispersion coefficients (logP and logS); the number of hydrogen bonds was counted from docking-based results. Overall, all the candidates well satisfy Lipinski's criteria for the argument on drug-likeness, ie: molecular mass < 300 amu; hydrogen-like donors < 5; hydrogen-like acceptors < 5; partition coefficient logP < 0. From the view of biological compatibility,
Physicochemical Properties of Studied Compounds

counted from docking results of each most effective ligand-PTP1B inhibitory complex.
Pharmacological Properties
The ADMET properties of the compounds are summarized in Table 5. The thresholds for theoretical prediction are based on Pires’ interpretations.
61
Regarding absorption, all the candidates register moderate intestinal absorbability (ca. 40%) and low Caco2 permeability (logPapp <
ADMET-based pharmacokinetics and pharmacology of the studied compounds

Discussion
Regarding the significant findings, this work preliminarily argue for the insights on the anti-diabetic potentiality of Aruncus dioicus. The isolated components, ie sambunigrin (
Regarding the experimental settings, the methanol extract from the A. dioicus leaf underwent partitioning to yield ethyl acetate and butanol fractions. Overall, the latter was preliminarily tested for overall inhibitory effect on PTP1B assay with a promising result (over 50% inhibition at concentration 30 μg.mL−1), thus herein subjected for chromatographic isolation and spectroscopic characterization obtaining four alkaloidal compounds (
Regarding the computational limitations, the computer-based platforms this work were utilized for their cost and time advantages given the purpose of wide-scale screenings. After specifying the most promising candidates, the more power-intensive technique, ie molecular dynamics simulation, is recommended for further consideration. This can monitor the real-time behavior of all the concerned ligand-protein atoms, thus deriving the inhibitory mechanism more particularly. However, it is noteworthy that the computer power required for this in-depth analysis is considerably intensive, thus not highly reasonable at screening and discovering stages.
Conclusions
This study first-time investigates the anti-diabetic potentials of glycosidic alkaloidal compounds isolated from Quan Lan Island A. dioicus aerial parts by a thoroughgoing investigation, from theoretical screening to experimental work. Firstly, experimental isolation and spectroscopic characterization identified for compounds, ie sambunigrin (
Methodology
Experimental Material Preparation
The aerial part of A. dioicus was collected in June 2021 from Quan Lan island, Quang Ninh province, Vietnam. The sample was sent for botanical identification by Dr Nguyen Quoc Binh, and a voucher specimen (AD01QL) was sent for formal deposit at the Institute of Natural Products Chemistry (INPC), Vietnam Academy of Science and Technology (VAST).
Extraction, Isolation, and Purification
The aerial parts of A. dioicus were chopped, washed, and dried (temperature appx. 50 °C) to reach desirable moisture (ca. 12%). Afterwards, a dried sample was grounded into powder (particle size appx. 0.2 mm; quantity 2 kg), before subjected to two consecutive extractions using ethyl acetate (EtOAc) at the feed-solvent ratio 1/3 w/v (ultrasonic assistance; temperature 35 °C; duration 90 min). Subsequently, the solvent was evaporated; the remaining powder residue underwent further extraction with methanol at the feed-solvent ratio 1/5 w/v (ultrasonic assistance; temperature 40 °C; duration 120 min). The extract obtained was subsequently filtered and the solvent was removed through vacuum distillation, yielding a total methanol extract (145 g).
The total methanol extract was then transferred into a standard silica gel chromatography column (particle sizes 63-200 μm). Elution was carried out using a solvent system CH2C2/MeOH. Initially, elution was performed with a polarity ratio of 10/1 v/v. Afterwards, the value was gradually increased to 1/10 v/v. This multi-step process yielded 15 fractions, denoted as AD.1-AD.15.
The certain fractions were selected for purification. Firstly, fraction AD.12 was further subjected to chromatography on an RP-C18 column (column size 4.0 × 60 cm; particle size 150 μm). A stepwise gradient of MeOH-H2O (from 1:4 to 2:1) was employed; this resulted in six subfractions denoted as AD.12-1 - AD.12-6. Subfraction AD.12-3 was continuously subjected to further purification using semi-preparative Agilent HPLC 1260 by an isocratic solvent system (25% MeOH in H2O; duration 40 min); this yielded compounds
Spectroscopic Characterization
Sambunigrin (
Prunasin (
Uridine (
Adenosine (
The chemical formulae of the characterized compounds (

Chemical structure of isolated compounds (
PTP1B in Vitro Assay
The inhibitory activity of isolated compounds (
The reaction kinetics (inhibition mode) was studied using the Lineweaver and Burk (double-reciprocal) plot method (GraphPad PRISM version 5, USA). The experiments were conducted using the similar procedure used in the PTP1B assay at various concentrations of compounds 
α-Glucosidase in Vitro Assay
The assay to assess the inhibitory activity of the α-glucosidase enzyme was conducted utilizing a 96-well microplate. The reaction components include p-nitrophenyl α-D-glucopyranoside (2.5 mM), phosphate buffer (100 mM; pH 6.8), α-glucosidase (0.2 U/mL), and the test samples (10 µL) at different concentrations; the total volume of each reaction mixture was 200 µL. In negative control lines, the test sample was replaced with a phosphate buffer (100 mM); while, acarbose was used as a positive control. The mixtures were incubated (temperature 37 °C; duration 30 min); afterwards, the reaction was stopped at the addition of Na2CO3 solutions (0.1 M). The absorbance of each product mixture was measured (wavelength 405 nm) using Infinite F50 spectrophotometer (Tecan, Switzerland). All tests are in triplicate. The percentage inhibition of α-glucosidase activity (%I) was calculated using the following expression: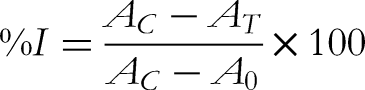
The IC50 values were calculated based on the log(concentration of test sample) and % inhibition curve, which represent the concentration of the test samples that inhibits 50% of α-glucosidase enzyme activity.
Statistical Analysis
The data were expressed as the mean (± standard deviation) from a minimum of three independent experiments (each performed in triplicate assays), and determined through regression analysis. SigmaPlot (version 11.0) was used for the statistical analysis.
Computational Input Preparation
The data is from experimental findings in this study and the existing literature serves as the input for the computational screening. Figure 7 presents the chemical formulae of potential compounds (

Biological assemblies of PTP1B, 3W37, 4W93, and structural formula of controlled drugs (ursolic acid -
Docking Simulation
A typical procedure of molecular docking simulation (by MOE 2012.10 63 ) follows three steps, ie: (i) Input preparation (configuration: protein active range 4.5 Å, ligand charge-assigning using Gasteiger-Huckel method); (ii) Docking simulation (configuration: retaining poses 10; solutions per iteration 1000; solutions per fragmentation 200); (iii) Re-docking iteration (threshold: root-mean-square deviation (RMSD) values < 2 Å). Given theoretical interpretation, the inhibitory effectiveness of a ligand towards the targeted protein structure can be primarily evaluated by docking score (DS) energy of the associated inhibitory system, which represents pseudo-Gibbs free energy (formed by hydrophilic binding and hydrophobic interaction); also, RMSD values and number of hydrogen-like interactions can be considered for arguments on bio-conformational rigidity and binding strength, respectively. In addition, MOE can provide visual rendering for ligand-protein interaction maps (2D) and in-pose arrangements (3D).
Quantum Calculation
Molecular chemical properties of the investigated structures were given by density functional theory (DFT) calculation using Gaussian 09 without symmetry constraints. 64 Level of theory M052X/6–311++G(d,p) and basis set def2-TZVPP 65 were selected. The converged geometries were checked for the structural global minimum on the potential energy surface (PES) by vibrational frequencies. The frozen-core approximation for non-valence-shell electrons was applied. The resolution-of-identity (RI) approximation was set. The frontier orbital analysis was carried out by NBO 5.1 at the level of theory M052X/def2-TZVPP. 66
Physicochemical Analysis
A combination model was used to predict the drug-like properties of the phytochemicals. The parameters were the physical properties retrieved from QSARIS 67 (based on Gasteiger–Marsili method 68 ). The references were from Lipinski's rule of five, 69 which provides the theoretical criteria for a well membrane-permeable candidate, ie: molecular mass < 500 Da; hydrogen-bond donors ≤ 5; hydrogen-bond acceptors ≤ 10; logP < + 5.70,71
ADMET Analysis
A combination model was used to predict the pharmacological potentiality of the compounds. The parameters were ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties retrieved from SwissADME (Swiss Institute of Bioinformatics; http://www.swissadme.ch/; 30th October 2023). The references were from Pires’ theoretical interpretations. 61
Supplemental Material
sj-docx-1-npx-10.1177_1934578X241271648 - Supplemental material for Inhibitory activities of Aruncus dioicus alkaloidal glycosides against protein tyrosine phosphatase 1B and α-glucosidase: A methodical theory-experiment investigation
Supplemental material, sj-docx-1-npx-10.1177_1934578X241271648 for Inhibitory activities of Aruncus dioicus alkaloidal glycosides against protein tyrosine phosphatase 1B and α-glucosidase: A methodical theory-experiment investigation by Phi-Hung Nguyen, Thanh Q. Bui, Thi-Tuyen Tran, Thi-Thuc Bui, Thi-Thuy Do, Dao-Cuong To, Manh Hung Tran, Phan Tu Quy, Nguyen Quang Co, Nguyen Vinh Phu and Nguyen Thi Ai Nhung in Natural Product Communications
Footnotes
Acknowledgements
Nguyen Thi Ai Nhung acknowledges the partial support of Hue University under the Core Research Program [Grant Number: NCTB.DHH.2024.04]
Authorship Contributions
All authors contributed to the study conception and design. All authors read and approved the final manuscript. Specific roles are summarized in the follow-up table.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical Approval is not applicable for this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Vietnam Academy of Science and Technology with project [Grant number: VAST04.05/22-23].
Supporting Information
In-detail spectra for experimental characterization provided in supporting information.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.