
Abstract
Introduction
Vitiligo is a prevalent, circumscribed hypopigmented skin ailment, affecting approximately 1% to 2% of the global populace. 1 It predominantly manifests on facial, cervical, extremity, abdominal, dorsal, and other regions rich in sweat glands. 2 The primary causative factors encompass oxidative stress, genetic predisposition, autoimmunity, neurohumoral influences, and self-destruction of melanocytes. 3 Notably, oxidative stress theory assumes a pivotal role in both the onset and progression of vitiligo. 4
Oxidative stress denotes the perturbation of the redox equilibrium, wherein the generation of reactive oxygen species (ROS) surpasses their utilization. The resultant surplus of ROS inflicts damage upon macromolecules integral to cellular processes, thereby eliciting cellular and tissue detriment, culminating in disease manifestation. 5 An expanding body of research underscores the role of human immortalized epidermal cells (HaCaT cells) as mediators linking oxidative stress to autoimmunity, particularly in the apoptotic cascade of melanocytes. 6
Constituting approximately 80% of cells in the human epidermis, HaCaT cells intricately interface with melanocytes both structurally and functionally, thereby exerting influence over the processes of proliferation, differentiation, and apoptosis of melanocytes.7‐8 Melanin synthesis initiates within melanocytes in the basal layer of the epidermis, subsequently translocating to adjacent HaCaT cells in the form of melanin vesicles. Ultimately, secretion occurs as HaCaT cells migrate from the basal layer to the stratum corneum, eventually shedding. 9 HaCaT cells wield their secreted growth factors and cellular matrix components to modulate melanocyte proliferation, melanin synthesis, and transport activities. 10
While melanocytes are the focal point in vitiligo research, HaCaT cells play an indispensable role that often goes unnoticed. Recognizing their significance could offer novel insights into vitiligo pathogenesis and therapeutic interventions.
Vernonia anthelmintica (L.) Willd, deeply rooted in Chinese traditional botanical medicine with historical mentions dating back to ancient times, 11 stands as an annual herb within the Vernonia genus of the Asteraceae family. Documented in the “Flora of China,” this botanical marvel is indigenous to Xinjiang and is renowned for its distinctive role in treating vitiligo. Its versatile therapeutic properties include dispelling wind, eliminating blood stasis, enhancing blood circulation, insecticidal actions, heat detoxification, and addressing dampness, swelling, cold, and pain relief.12‐13
The crude extract and isolated compounds derived from V anthelmintica exhibit a myriad of pharmacological activities. These encompass antivitiligo, antipsoriasis, antioxidant, antidiabetic, anti-inflammatory, hepatoprotective, neuroprotective, analgesic, antipyretic, antiparasitic, antibacterial, immunomodulatory effects, and promotion of estrogen synthesis. 14 Notably, the anthelmintic injection, employed since the 1970s for vitiligo treatment, has demonstrated efficacy in promoting repigmentation. 15
Research highlights flavonoids and caffeic acid compounds as the pivotal active ingredients in V anthelmintica for vitiligo treatment, with flavonoids being the focal point of investigation. 16 In this study, we extracted specific flavonoids—Butin, Butein, Eriodictyol, and Liquiritigenin—from V anthelmintica seeds. Our experimentation on mouse melanoma cells (B16) focused on evaluating their impact on tyrosinase activity and melanin content. Previous findings indicated that these flavonoids activate tyrosinase and elevate melanin content within a specific concentration range in mouse B16 cells. Building upon this groundwork, we investigate the effects of these flavonoids on H2O2-induced oxidative stress injury in HaCaT cells.
The chemical structural formulae of the four flavonoids under scrutiny are elucidated in Figure 1.

Chemical structure formula of the four flavonoids.
The nuclear transcription-related factor (Nrf2) and antioxidant response element (ARE) system stands as a pivotal defensive mechanism combating oxidative stress, intricately involved in the pathogenesis and progression of various diseases. 17 This system responds to exogenous stimuli and oxidative stress, orchestrating cellular responses. In the absence of stimuli, 4 Nrf2 remains sequestered in the cytosol, forming complexes with the chaperone protein Keap1, thereby maintaining a subdued state. However, under conditions of oxidative stress, Nrf2 disengages from Keap1, translocates to the nucleus, and binds to AREs. This nuclear interaction activates a cascade, prompting the expression of phase II detoxification enzymes and antioxidant genes.
The experiment at hand seeks to establish an oxidative stress model in HaCaT cells induced by hydrogen peroxide. Through this model, we aim to assess the impact of four flavonoids on malondialdehyde (MDA) content, superoxide dismutase (SOD) activity, 4 Nrf2 protein expression, and downstream antioxidant gene expression. These measurements are instrumental in discerning the flavonoids’ effects on the oxidative stress milieu within HaCaT cells challenged by hydrogen peroxide. Our goal is to unravel the protective mechanisms against cellular oxidative stress damage. This investigation contributes valuable insights into the theoretical framework for employing flavonoids from V anthelmintica (L.) Willd in vitiligo treatment and the development of more efficacious drugs.
Materials and Methods
Materials and Reagents
HaCaT cells, procured from the Shanghai Cell Bank, Chinese Academy of Sciences, served as the cellular substrate for this study. MEM medium (Procell Life Science & Technology Co. Ltd) provided the essential cell culture milieu, while DMSO (Sigma), Penicillin-Streptomycin Solution (BI), fetal bovine serum (BI), Trypsin EDTA solution (BI), and PBS buffer (BI) were utilized for cell maintenance and manipulation. Key biochemical assays were facilitated by the MTT kit, BCA protein quantification kit, RIPA buffer, and PMSF (Beijing Solarbio Science & Technology Co. Ltd). The SOD kit and MDA kit were procured from Nanjing Jiancheng Bioengineering Research Institut.
Chemical reagents, including 30% hydrogen peroxide (Xi'an Chemical Reagent Factory), glacial acetic acid (Xi'an Chemical Reagent Factory), Trizol (Ambion), trichloromethane, isopropyl alcohol, and anhydrous ethanol (Chengdu Kelong Chemical Co. Ltd), were employed for various experimental procedures. DEPC water (Beijing Solarbio Science & Technology Co. Ltd) ensured purity in molecular biology applications. Essential lab consumables, such as DNase/RNase free tips, DNase/RNase free tubes, and polymerase chain reaction (PCR) Strips with 8 wells, were sourced from Shanghai Kejin Biotechnology Co. Ltd.
Molecular biology assays were conducted using a cDNA reverse transcription kit and fluorescent quantitative PCR kit (Takara). Sodium dodecyl sulfate (SDS)-PAGE Separating Gel Buffer (1.5 M Tris-HCL pH8.8, 0.4% SDS) and SDS-PAGE Stacking Gel Buffer (1 M Tris-HCL pH6.8, 0.4% SDS), along with 20× TBST Buffer, were obtained from Beijing Solarbio Science & Technology Co. Ltd For protein transfer, polyvinylidene fluoride (PVDF) membrane (Millipore) was employed.
Antibodies, including Nrf2, Heme oxidase-1 (HO-1), and NQO1 antibodies (Proteintech) and GAPDH antibody (Abcam), were utilized for Western blot analyses. The ECL Chemiluminescence Kit was procured from Beijing Labgic Technology Co. Ltd Flavonoids Butin and Butein (purity ≥ 98%, HPLC analysis, Shanghai Yuanye Bio-Technology Co. Ltd) and Eriodictyol and Liquiritigenin (purity ≥ 98%, HPLC analysis, Beijing Solarbio Science & Technology Co. Ltd) were acquired for experimental use.
Main Instruments and Equipment
Key laboratory apparatus used in this study included a Cell Culture Incubator (SANYO), an Ultra Clean Bench (Sujing Group Suzhou AIR TECH Technology Co. Ltd), an 80 °C Refrigerator (Panasonic), Liquid Nitrogen Biological Containers (Chengdu Liquid Ammonia Containers Factory), an Enzyme Labeler (Thermo), an Ultra-micro Nucleic Acid Analyzer (Hangzhou Aosheng Instrument Co. Ltd), a Fluorescence Quantitative PCR Instrument (Hangzhou Bioer Technology Co. Ltd), a Protein Electrophoresis Instrument (Bio-Rad), and an E-BLOT Contact Chemiluminescence Imaging System (Anhui Huite Medical Technology Development Co. Ltd).
Cell Culture
HaCaT cells were brought out from the liquid nitrogen container, lysed immediately in a 37 °C water bath, removed from the freezing solution and suspended in fresh medium. Subsequently, the cells were inoculated in a culture flask with complete MEM medium containing 15% fetal bovine serum and 1% Penicillin–Streptomycin Solution, followed by incubation at 37 °C in a 5% CO2 cell incubator. After 24 h, the cells were observed using an inverted microscope and periodically changed the ulturing medium with the fresh complete MEM medium. When the density of adherent cells reached 80% to 95%, they were passaged. Cells of the logarithmic growth phase were selected for the experiment. The experiments were grouped as follows:
Control: HaCaT cells were cultured without the addition of drugs or hydrogen peroxide in a cell culture incubator containing 5% CO2 at 37 °C.
H2O2 Induction Model: Cells received no dosing but were subjected to 600 μmol/L H2O2 treatment for 12 h. 18
Drug Treatment: HaCaT cells were pretreated with 6.25, 12.5, and 25 µmol/L of Butin, Butein, Eriodictyol, and Liquiritigenin, respectively. Subsequently, they were incubated in an incubator containing 5% CO2 at 37 °C for 24 h. Following this, the cells were treated with 600 µmol/L H2O2 and continued to be incubated in an incubator containing 5% CO2 at 37 °C for 12 h.
Cytotoxicity Assay of Four Flavonoids and Their Impact on the Proliferation of H2O2-Induced HaCaT Cells Using the MTT Method
Cells were seeded in 96-well plates at a density of 1 × 104 cells/well, with six replicate wells allocated for each group. Following incubation, the control and model groups underwent a liquid exchange, while the experimental groups were treated with varying concentrations (1.563, 3.125, 6.25, 12.5, 25, 50, 100 µmol/L) of Butin, Butein, Eriodictyol, and Liquiritigenin for 24 h. After this incubation period, 100 µL of MTT solution (5 mg/mL) was added to assess the cytotoxicity of the four compounds.
For pretreatment, cells were again seeded as described above and exposed to the drugs for 24 h. Subsequently, the old culture medium was replaced, and the cells were treated with 600 µmol/L H2O2 for 12 h. After H2O2 exposure, the old culture medium was discarded, and cells were washed once with PBS. The impact of the four flavonoids on HaCaT cell proliferation induced by H2O2 was evaluated by adding 100 µL of MTT solution (5 mg/mL) to each well. The supernatant was discarded, and 100 µL of DMSO was added to each well, followed by shaking for 10 min to completely dissolve the formazan crystals. The cell proliferation rate or inhibition rate was calculated, and the IC50 value was determined using SPSS 27 software.


Measurement of Oxidative Stress-Related Indicators
Intracellular SOD Activity Assay
The SOD activity was assessed using the WST-1 method. Initially, cell samples were prepared by seeding 1 × 106 cells/well in six-well plates. After cell adherence, HaCaT cells underwent pretreatment with Butin, Butein, Eriodictyol, and Liquiritigenin at concentrations of 6.25, 12.5, and 25 µmol/L, respectively, for 24 h. Each sample group comprised three replicate wells. Subsequently, the cells were exposed to 600 µmol/L H2O2 for 12 h.
Next, the cells were harvested, and RIPA lysate was employed to lyse the cells and the concentration of sample protein was determined following the BCA kit instructions. Finally, the test samples, enzyme working solution, enzyme diluent, substrate application solution, and other reagents were meticulously combined in a 96-well plate as per the SOD kit instructions. The mixture was incubated at 37 °C for 20 min, and the optical density value was measured at 450 nm using an enzyme marker. The calculation formula is:



Determination of Intracellular MDA Content
The MDA content was assessed using the thiobarbituric acid method. The protocol for preparing and collecting cell samples was consistent with that described in 1.5.1. Subsequent steps followed the guidelines provided by Nanjing Jiancheng Biotechnology for the determination of MDA content. The calculation formula is:

Detection of Intracellular Expression of Antioxidant-Related Genes by Fluorescent Quantitative PCR
Total cellular RNA was extracted using the Trizol method. Subsequent cDNA synthesis was carried out with the Prime Script TM RT reagent Kit with gDNA Eraser (Perfect real time) kit, strictly following the provided instructions. Real-Time PCR (RT-PCR) analysis employed the TB Green® Premix Ex TaqTM II (Tli RNaseH Plus) kit for detecting the expression of relevant mRNAs. The 2−ΔΔCt method was employed for calculation, and the data were analyzed using GAPDH as an internal reference control. All primers were synthesized by Sangon Biotech (Shanghai) Co. Ltd, with the sequences listed in Table 1.
Primer Sequences for Real-Time PCR.

Abbreviations: PCR, polymerase chain reaction; HO-1, heme oxidase-1; Nrf2, nuclear transcription-related factor; NQO1, quinone oxidoreductase.
Western Blotting
HaCaT cells underwent treatment according to the experimental grouping. Protein extraction followed established literature procedures. Protein content was quantified using the BCA protein kit, and sample protein concentrations were adjusted to a uniform level. Each sample contributed 20 μg of protein, added to SDS sample buffer, and proteins were separated through 10% polyacrylamide gel electrophoresis. Postelectrotransfer to PVDF membranes, these membranes underwent blocking with 5% skim milk powder for 1 to 2 h at room temperature. After three washes with TBST, the membranes were incubated overnight at 4 °C with primary antibodies, including Nrf2 (1:1000 dilution), NQO-1 (1:10000 dilution), HO-1 (1:1000 dilution), and GAPDH (1:1000 dilution), followed by slow agitation. Subsequently, after washing, the membranes were exposed to horseradish peroxidase-conjugated secondary antibodies at a dilution of 1:10000 for 1 h at room temperature. The chemiluminescence reaction utilized the ECL Chemiluminescence Kit, and band observation occurred with the E-BLOT contact chemiluminescence imaging system. ImageJ software was employed for light grayscale value analysis. The presented data represent at least 3 experiments.
Statistical Analysis
Every experiment underwent replication a minimum of three times. Experimental data were expressed as mean ± standard deviation (SD). One-way ANOVA was employed for intergroup comparisons, with P < .05 indicating significance and P < .01 indicating high significance. Statistical analysis and graphing were performed using GraphPad Prism8 software.
Results
Cytotoxicity Experiments of Four Flavonoids and their Impact on the Proliferation of H2O2-Induced HaCaT Cells
As depicted in Figure 2, the four flavonoids exhibited no cytotoxicity and, at lower concentrations, facilitated the proliferation of HaCaT cells. However, escalating drug concentrations led to observable cytotoxic effects.

Cytotoxicity of four flavonoids. Note: (a) Treated with Butin, (b) treated with Butein, (c) treated with Eriodictyol, (d) treated with Liquiritigenin. Compared with control, ns. P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001.
Following the administration of varying doses of the four flavonoids (1.563, 3.125, 6.25, 12.5, 25, 50, and 100 µmol/L) for 24 h, the cells underwent induction with 600 µmol/L H2O2 for an additional 12 h. The control group (CT) signifies cells without drug intervention but with the addition of H2O2. The impact of the four flavonoids on H2O2-induced HaCaT cell proliferation was evaluated using the MTT kit. Figure 3 illustrates that the flavonoids hindered cell proliferation in a concentration-dependent manner. Subsequently, concentrations of 6.25, 12.5, and 25 µmol/L of Butin, Butein, Eriodictyol, and Liquiritigenin were selected for subsequent experiments.

Effect of flavonoids on the proliferation rate of H2O2-induced HaCaT cells. Note: (a) Treated with Butin, (b) treated with Butein, (c) treated with Eriodictyol, (d) treated with Liquiritigenin. Compared with control, ns. P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001.
Measurement of Oxidative Stress-Related Indicators
Effect of Four Flavonoids on H2O2-Induced SOD Activity in HaCaT Cells
To assess the antioxidant potential of the four flavonoids, intracellular SOD activity was measured. As illustrated in Figure 4, the SOD activity in the model group was notably lower than that in the control (P < .0001), exhibiting an approximately 41.23% decrease. Conversely, the medium and high-dose drug-treated groups exhibited a gradual and significant increase in SOD activity (P < .01) with rising Butin content. The high-dose Butin-treated group (S 25) demonstrated the highest SOD activity at 211.54 U/mg prot, marking a 38.27% increase compared to the model. Similar trends were observed in the Butein-treated group, with the high dose (Y 25) displaying the highest SOD activity at 213.35 U/mg prot, a 39.92% increase over the model.

Effects of four flavonoids on H2O2-induced intracellular superoxide dismutase (SOD) activity. Note: (a) Treated with Butin, (b) Treated with Butein, (c) Treated with eriodictyol, (d) Treated with liquiritigenin. H2O2 model compared with CT: ns. P > 0.05, #P < .05, ##P < .01, ###P < .001, ####P < .0001. Drug-treated group compared with H2O2 model: ns. P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001.
Comparatively, the SOD activity in the Eriodictyol-treated groups (L, M, and H) gradually and significantly increased (P < .05 or P < .001) with escalating concentrations. The high-dose Eriodictyol-treated group (ER 25) exhibited the highest SOD activity at 238.07 U/mg prot, representing a remarkable 56.13% surge compared to the model. Furthermore, the Liquiritigenin-treated group showcased significantly elevated SOD activity compared to the model (P < .01 or P < .001), with the high-dose Liquiritigenin-treated group (LI 25) reaching the highest SOD activity at 227.93 U/mg prot, indicating a 49.48% increment compared to the model.
In summary, intracellular SOD activity experienced a substantial decline under H2O2 induction. Notably, all four flavonoids exhibited a dose-dependent enhancement in SOD activity, with the Eriodictyol-treated group demonstrating the highest efficacy (Figure 4c), showcasing superior cellular protection against oxidative stress damage.
Effect of Four Flavonoids on H2O2-Induced MDA Content in HaCaT Cells
To delve deeper into the antioxidant potential of the four flavonoids, intracellular MDA content was assessed. Comparative analysis with the control revealed a significant elevation (P < .001) in intracellular oxidative damage marker, MDA, following H2O2 induction of cells. However, after treatment with flavonoids including Butin, Butein, Eriodictyol, and Liquiritigenin, a marked reduction in MDA content was observed, as depicted in Figure 5.
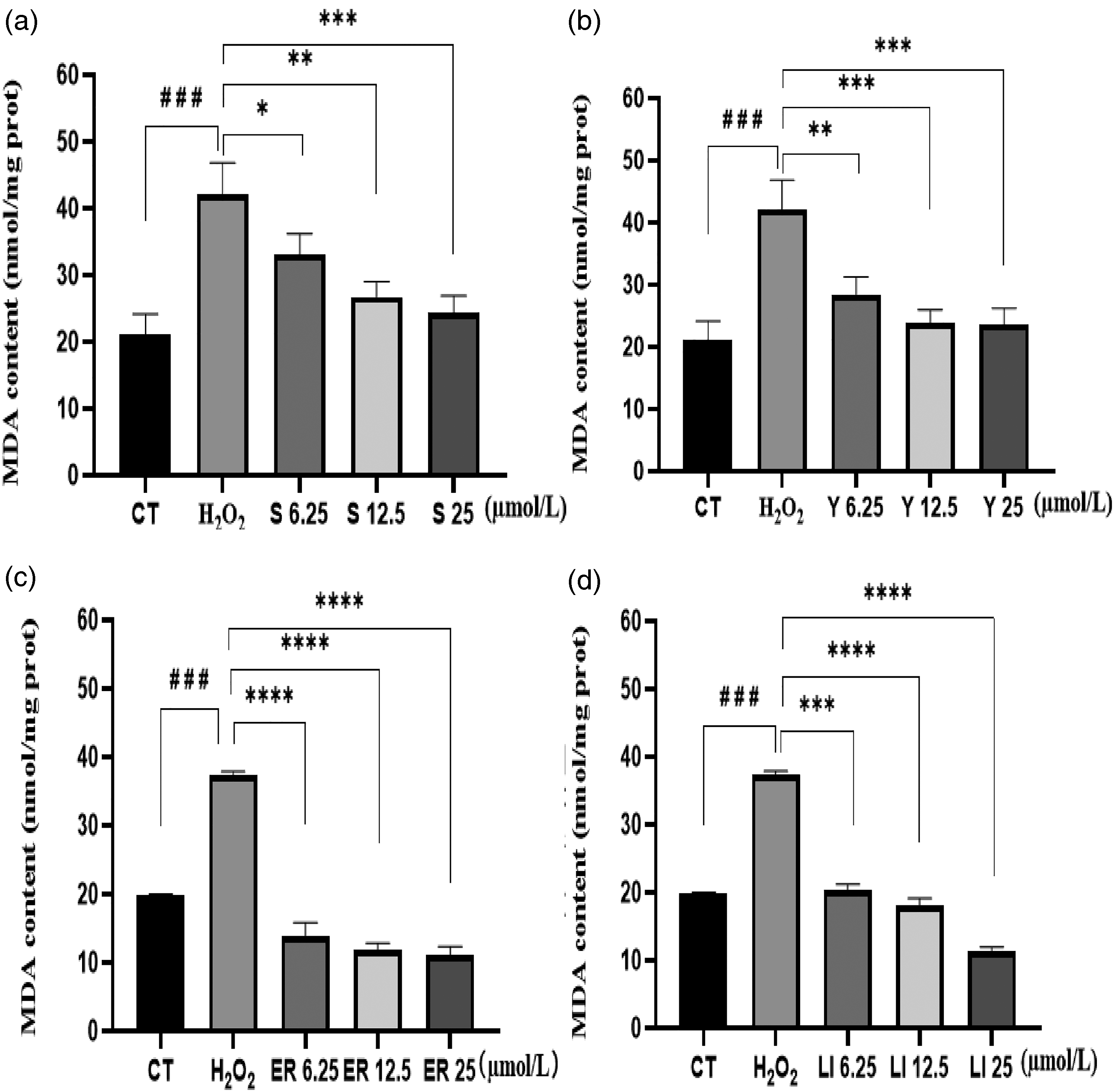
Effects of four flavonoids on H2O2-induced intracellular malondialdehyde (MDA) content. Note: (a) Treated with Butin, (b) treated with Butein, (c) treated with Eriodictyol, (d) treated with Liquiritigenin. H2O2 model compared with CT: ns. P > .05, #P < .05, ##P < .01, ###P < .001, ####P < .0001. Drug-treated group compared with H2O2 model: ns. P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001.
The MDA content exhibited a progressive decline with escalating drug concentrations, signifying a dose-dependent response to flavonoid treatment. Notably, all four flavonoids demonstrated the capability to decrease MDA content in a concentration-dependent manner. Among them, the Eriodictyol drug-treated group (Figure 5c) exhibited the lowest MDA content. At the highest drug dose, MDA content reached 11.52 nmol/mg prot, showcasing a substantial 68.86% reduction compared to the model (P < .0001). This highlights the superior ability of Eriodictyol to protect cells from oxidative stress damage.
Effects of Four Flavonoids on the Expression of Nrf2 and its Downstream Antioxidant Genes Such as HO-1 and NQO1
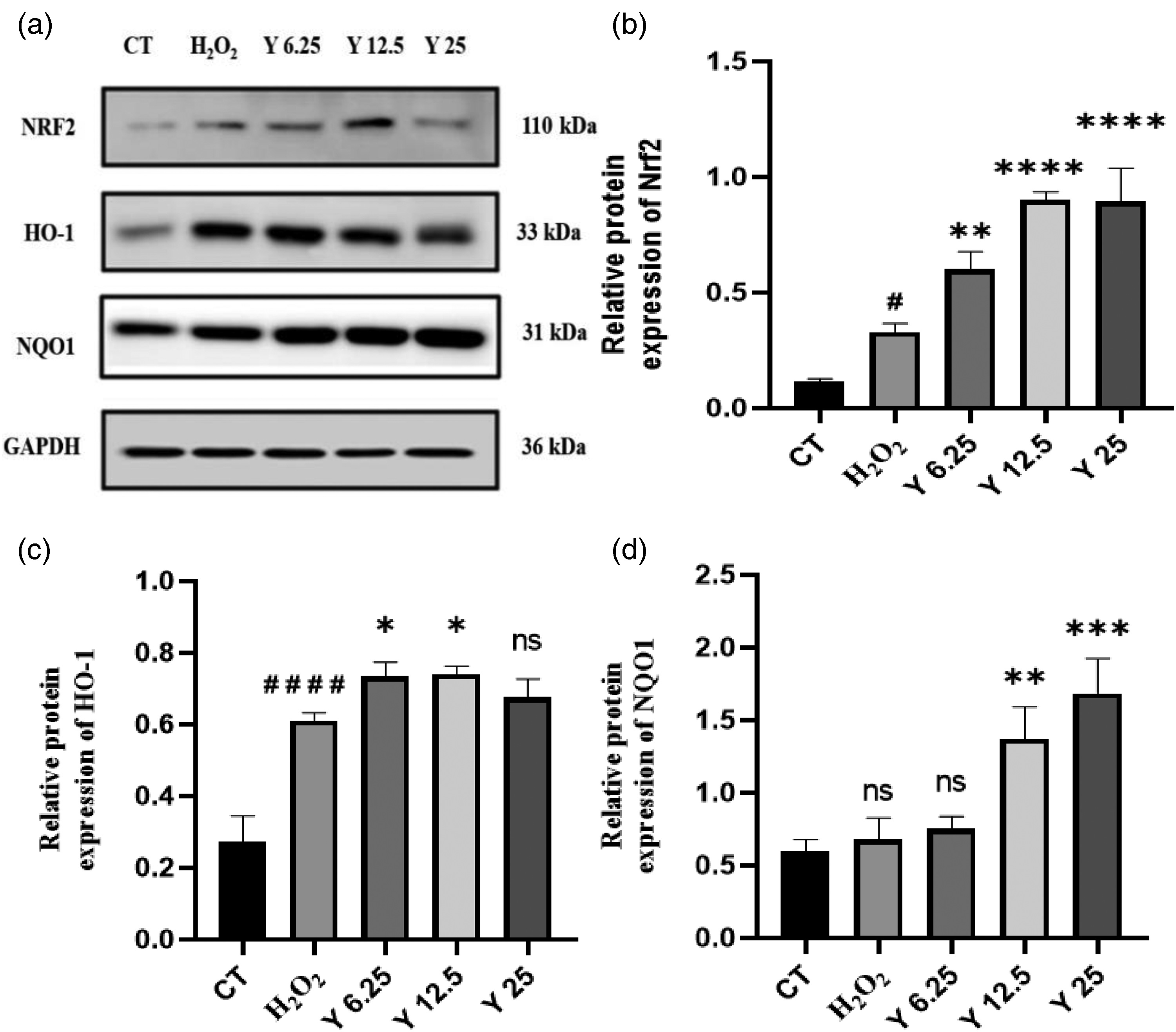
Quantitative RT-PCR was employed to investigate the expression of Nrf2-related genes and their downstream antioxidant genes, HO-1 and NQO1. Results depicted in Figures 6‐9 revealed that all four flavonoids exhibited the ability to enhance the expression of Nrf2, HO-1, and NQO1 mRNA.

Effect of butin treatment on relative mRNA expression in H2O2-induced HaCaT cells. Note: Drug-treated group compared with H2O2 model: ns. P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001.

Effect of butein treatment on relative mRNA expression in H2O2-induced HaCaT cells. Note: Drug-treated group compared with H2O2 model: ns. P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001.

Effect of eriodictyol treatment on relative mRNA expression in H2O2-induced HaCaT cells. Note: Drug-treated group compared with H2O2 model: ns. P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001.

Effect of liquiritigenin treatment on relative mRNA expression in H2O2-induced HaCaT cells. Note: Drug-treated group compared with H2O2 model: ns. P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001.
Upon closer examination of the figures, it is evident that the (H2O2-induced) Nrf2, HO-1, and NQO1 relative mRNA expression in the model displayed an upregulation compared with the control, though statistically nonsignificant (P > .05). At low concentrations of drug administration (6.25 µmol/L), there was no statistical difference in the relative mRNA expressions of Nrf2, HO-1, and NQO1 (P > .05). However, with increasing concentrations of Butin, Butein, Eriodictyol, and Liquiritigenin, there was a significant upregulation in the relative mRNA expressions of these genes.
In the Butin drug-treated group (Figure 6), Nrf2, HO-1, and NQO1 relative mRNA expression demonstrated a highly significant upregulation (P < .01, P < .001, or P < .0001) at Butin concentrations of 12.5 and 25 µmol/L compared with the model. Similar trends were observed in the Butein drug-treated group (Figure 7), where the relative mRNA expression of Nrf2, HO-1, and NQO1 exhibited a highly significant upregulation at a Butein concentration of 25 µmol/L compared with the model group (P < .01 or P < .0001).
In the Eriodictyol drug treatment group (Figure 8), the expression of Nrf2, HO-1, and NQO1 relative mRNA increased in a dose-dependent manner. HO-1 relative mRNA expression was highly significantly upregulated at all three drug concentrations compared with the model (P < .01 or P < .0001). Nrf2 and NQO1 expression at an Eriodictyol concentration of 6.25 µmol/L were not statistically different (P > .05), mRNA expression was significantly up-regulated (P < .05) at a concentration of 12.5 µmol/L, and mRNA expression was highly significantly up-regulated (P < .001) at a concentration of 25 µmol/L.
In the Liquiritigenin drug-treated group (Figure 9), Nrf2 relative expression was significantly upregulated at the highest Liquiritigenin concentration compared with the model (P < .05). HO-1 relative expression was highly significantly upregulated at both Liquiritigenin concentrations of 12.5 and 25 µmol/L (P < .01 or P < .0001), and NQO1 relative expression was only significantly upregulated at Liquiritigenin concentration of 12.5 µmol/L (P < .05).
These findings collectively suggest that Butin, Butein, Eriodictyol, and Liquiritigenin can effectively shield cells from oxidative stress damage by upregulating the expression of antioxidant genes Nrf2, HO-1, and NQO1.
Effects of Four Flavonoids on the Expression of Nrf2 and its Downstream Antioxidant Proteins Such as HO-1 and NQO1
The expression of Nrf2-associated antioxidant proteins was scrutinized via Western blotting, and the findings are presented in Figures 10‐13. Notably, Nrf2, HO-1, and NQO1 protein expressions exhibited an upregulation in the model when compared with the control. In the experimental groups, a dose-dependent up-regulation of Nrf2, HO-1, and NQO1 proteins was observed with increasing concentrations of Butin, Butein, and Eriodictyol; however, a down-regulation was noted at 25 µmol/L in the case of Liquiritigenin.

Upregulation of Nrf2 in butin-treated HaCaT cells enhances H2O2-induced activation of Nrf2 and expression of downstream antioxidant genes HO-1, NQO-1. Note: (a) Treated with Butin, (b) treated with Butein, (c) treated with Eriodictyol, (d) treated with Liquiritigenin. H2O2 model compared with CT: ns. P > .05, # P < .05, ## P < .01, ### P < .001, #### P < .0001. Drug-treated group compared with H2O2 model: ns. P > .05, * P < .05, ** P < .01, *** P < .001, **** P < .0001. Abbreviations: Nrf2, nuclear transcription-related factor; HO-1, heme oxidase-1.
Upregulation of Nrf2 in Butein treated HaCaT cells enhances H2O2-induced activation of Nrf2 and expression of downstream antioxidant genes HO-1, NQO-1. Note: (a) treated with Butin, (b) treated with Butein, (c) treated with Eriodictyol, (d) treated with Liquiritigenin. H2O2 model compared with CT: ns. P > .05, #P < .05, ##P < .01, ###P < .001, ####P < .0001. Drug-treated group compared with H2O2 model: ns. P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001. Abbreviations: HO-1, heme oxidase-1; Nrf2, nuclear transcription-related factor.

Upregulation of Nrf2 in Eriodictyol treated HaCaT cells enhances H2O2-induced activation of Nrf2 and expression of downstream antioxidant genes HO-1, NQO-1. Note: (a) treated with Butin, (b) treated with Butein, (c) treated with eriodictyol, (d) treated with Liquiritigenin. H2O2 model compared with CT: ns. P > .05, #P < .05, ##P < .01, ###P < .001, ####P < .0001. Drug-treated group compared with H2O2 model: ns. P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001. Abbreviations: HO-1, heme oxidase-1; Nrf2, nuclear transcription-related factor.

Upregulation of Nrf2 in Liquiritigenin-treated HaCaT cells enhances H2O2-induced activation of Nrf2 and expression of downstream antioxidant genes HO-1, NQO-1. Note: (a) Treated with Butin, (b) treated with Butein, (c) treated with eriodictyol, (d) treated with Liquiritigenin. H2O2 model compared with CT: ns. P > .05, #P < .05, ##P < .01, ###P < .001, ####P < .0001. Drug-treated group compared with H2O2 model: ns. P > .05, *P < .05, **P < .01, ***P < .001, ****P < .0001. Abbreviations: HO-1, heme oxidase-1; Nrf2, nuclear transcription-related factor.
Eriodictyol, among the four flavonoids, displayed a more pronounced pharmacological effect, resulting in the highest expression of Nrf2 and downstream antioxidant proteins HO-1 and NQO1. Notably, the most significant activation of Nrf2 and the highest expression of HO-1 and NQO1 proteins were witnessed at the concentration of 25 µmol/L of Eriodictyol. These observations were highly significant (P < 0.01 or P < 0.0001) when compared with the model.
These outcomes collectively indicate that Butin, Butein, Eriodictyol, and Liquiritigenin possess the capability to activate Nrf2, facilitating its translocation into the nucleus to enhance the transcriptional activity of Nrf2 and consequently promoting the expression of downstream antioxidant proteins HO-1 and NQO1.
Discussion
In this study, H2O2-induced HaCaT cells were used to construct a model of oxidative stress injury to HaCaT cells at the white patches of vitiligo patients. Oxidative stress-related indicators, namely SOD and MDA, were chosen to evaluate the protective effects of four flavonoids (Butin, Butein, Eriodictyol, Liquiritigenin) against oxidative stress, primarily through the Nrf2 signaling pathway. The cytoprotective potential of these flavonoids was assessed by investigating their impact on the expression of HO-1, NQO1, SOD, and other related antioxidant enzymes. The findings revealed that all four flavonoids activated the Nrf2 metabolic pathway, upregulated the expression of HO-1 and NQO1 proteins, and subsequently enhanced the expression of antioxidant enzymes, ultimately reducing cellular oxidative stress. Thus, Butin, Butein, Eriodictyol, and Liquiritigenin demonstrated significant efficacy in safeguarding HaCaT cells from oxidative stress. This research establishes a foundational understanding of the anti-vitiligo potential of flavonoids in V anthelmintica (L.) Willd, specifically in the context of oxidative stress damage models. The insights gained hold significant implications for advancing vitiligo treatment and developing more potent drugs rooted in the flavonoids of V anthelmintica (L.) Willd.
Vitiligo patients exhibit reactive oxygen cluster accumulation in the epidermis, heightened H2O2 levels in HaCaT cells and melanocytes around depigmented regions, and diminished antioxidant enzyme levels in both epidermal tissue and serum. 19 Oxidative stress profoundly influences various biological processes, including the normal metabolism, proliferation, and differentiation of melanocytes. The reduction or absence of melanocytes and pigment granules in the vitiligo-affected epidermis is closely linked to the oxidative stress microenvironment. This stress not only impact melanocytes but also HaCaT cells, which typically produce and secrete cytokines such as Stem Cell Growth Factor (SCF) and Basic Fibroblast Growth Factor (bFGF) in healthy skin. These cytokines play a dual role by promoting melanocyte survival and melanin synthesis. However, under oxidative stress, increased expression of miR25 in HaCaT inhibits the production of SCF and bFGF, exacerbating melanocyte damage and impeding melanin synthesis. 20 Suo et al 21 demonstrated a significant reduction in intracellular SCF and bFGF secretion in healthy human keratinocyte line HaCaT induced with oxidative damage (PM2.5). This suggests that HaCaT cells, within the oxidative stress microenvironment associated with vitiligo, can influence melanocyte survival and function through altered paracrine secretion.
Oxidative stress emerges as a pivotal contributor to the initiation and progression of vitiligo. Consequently, counteracting oxidative stress stands out as a promising avenue to facilitate pigmentation recovery in the depigmented patches of vitiligo patients. An increasing number of researchers are directing their attention toward investigating drugs with antioxidant properties. It is established through studies that oxidative stress exacerbates vitiligo within the epidermis. However, a comprehensive exploration is imperative to ascertain whether oxidative stress alone can induce the onset of vitiligo. Antioxidant therapy emerges as a novel approach in the clinical treatment of vitiligo, showing significant potential for broad application. Anthelmintic flavonoids, such as those identified in this study, may emerge as efficacious agents for treating or retarding oxidative stress-induced skin damage. 21 Nevertheless, it is crucial to acknowledge the limitations of the current study, necessitating further experiments. Subsequent investigations could delve into the impact of the four flavonoids on cytokine secretion in HaCaT cells, validating findings through animal experiments to unravel the molecular mechanisms governing the antioxidant effects of these flavonoids in HaCaT cells. Establishing a co-culture system of HaCaT and melanocyte cells will further illuminate the specific intracellular signaling pathways regulating melanin synthesis. The role of HaCaT cells in the incidence and development of vitiligo, the involvement of exosomes and related components, their potential role in promoting melanosome synthesis and transport, as well as the cytokines they secrete, remains unclear and necessitates in-depth exploration. Another constraint lies in the low water solubility of these flavonoids, requiring future investigations to enhance their water solubility while preserving efficacy in animal experiments.
Conclusion
Nrf2 stands as a pivotal transcription factor, orchestrating cellular defense against oxidative damage by activating antioxidant enzyme genes. 22 The Nrf2-ARE signaling pathway serves as the principal cellular mechanism combating exogenous substances and oxidative harm. Activation of this pathway, triggered by oxidative stress, prompts the translocation of Nrf2 from the cytoplasm, where it is bound to the chaperone protein Keap1, into the nucleus. In the nucleus, Nrf2 binds to the ARE, initiating the transcription of downstream antioxidant genes and phase II detoxification enzymes. This cascade of events imparts potent antioxidant effects, reinstating oxidative-antioxidant homeostasis in vivo. 23 Thus, initiating the Nrf2-ARE signaling pathway activates the expression of downstream antioxidant genes such as HO-1 and NQO1, alongside their respective proteins.
In this study, H2O2-induced HaCaT cells were used to construct a model of oxidative stress injury to HaCaT cells at the white patches of vitiligo patients. Following treatment with the four flavonoids, SOD activity exhibited a significant increase compared to the H2O2 induction model (P < .01), while MDA content showed a marked decrease compared to the H2O2 induction model (P < .01). The augmentation of SOD activity and reduction in MDA content were dose-dependent. All 4 flavonoids upregulated the expression levels of Nrf2, HO-1, and NQO1 genes, as well as their corresponding proteins. The flavonoids derived from V anthelmintica (L.) Willd demonstrated a protective impact against H2O2-induced oxidative stress damage in HaCaT cells. A visual representation of the study's conclusion is depicted in Figure 14.

Graphical conclusion of this study.
Footnotes
Author Contribution
Guliqiehere Eibulayin contributed to the experiments, data processing and statistical analysis, and writing the manuscript. Nurgul Rahman and Jian hong Fu's contributed to providing project funding. Adila Tuerxuntayi's contributed to supervising the conduct of experiments and manuscript grammar checking.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research was financed by the Program of Scientific Research of Xinjiang Colleges and Universities (XJEDU2017S029); National Natural Science Foundation of China (32060528), “Tianshan Innovation Team Plan” of Xinjiang (2022D14004).