
Abstract
Keywords
Introduction
Derivatives of 3,7-diazabicyclo[3.3.1]nonane (bispidines 1 ) are similar to several classes of biologically active alkaloids, for example, lupanine and anagyrine, and considered as potential antiviral agents.2,3 These tertiary amides are known to be strongly dependent upon the conformation in their manifold properties, 4 including diverse aspects of their biological activity.5,6 However, after more than a century of structural investigations of bicyclo[3.3.1]nonane derivatives, 7 the principal factors governing their conformational behavior are not yet completely clear despite the relative simplicity of the underlying transformations. 8


Four skeletal conformers are commonly postulated for these molecules (Figure 1), named according to the forms of two 6-membered rings that comprised the bicyclic structure: “double chair,” or

3,7-dialkylbispidine skeletal conformations.
Recent theoretical investigations
8
characterized the
Some of the 3,7-diacyl bispidine derivatives are known to possess useful properties, tightly bound to the state of internal rotation for the acyl substituents.12,13 The
Considering the skeleton conformation to be invariant, the change in the relative acyl substituent orientation—parallel (

The “rigid double chair” pseudoatomic viewpoint on the internal rotation in 3,7-diacylbispidines.
The most common research techniques of the conformational analysis so far remain the experimental assessment of the state of conformational equilibria using the indirect observations of the conformational ratios from the NMR spectra and single-crystal X-ray diffraction structure determinations, that most often supplies us with the solid state conformer optimal structure. On the other hand, the whole conformation ensemble could be approximated using the structure and energy of forms obtained from the results of molecular modeling from first principles, using initially the minimal amount of the available experimental observations.
The present paper contains the detailed studies of potential energy surface (PES) minima of 3,7-diacylbispidines using the reliable high-level nonempirical correlated computational chemistry methods. Their possible changes in skeletal conformation were completely out of the scope of any computational studies due to the previous experimental findings. Actually, the coupling between the skeletal conformational interconversion and the carbonyl groups reorientation shall form the whole picture of the conformational behavior of the entitled compounds as in Figure 3, somewhat more complicated than reviewed above as based upon the experimental data. Here, we try to assess in greater detail (including all 6 possible spatial forms) the conformational behavior for 4 simplest 3,7-diacetylbispidines

The complete conformation coupling diagram for 3,7-diacylbispidines.


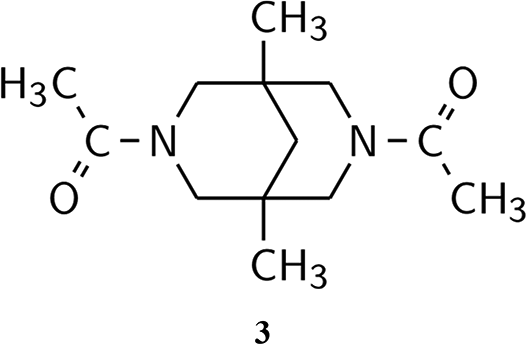

Methods
Conformational energy Econf(S) of a certain conformation of the molecular structure S is computed as the difference of the energy value for the investigated conformation versus that of the lowest energy (optimal) conformer of the molecule (S0) using either the absolute (optimized total, probably zero-point energy [ZPE]-corrected) energy E obtained from any kind of nonempirical calculations (1) or, similarly, using the molecular strain energy Eς (2):


Hereafter, we apply the bond separation reaction (BSR) formalism to estimate the strain of a molecular structure. Following the implicit axiom of chemistry that the molecule is a union of chemical bonds between atoms, we evaluate the strain energy of a structure as the energy effect of the formation reaction of the target structure S from strainless fragments. The formal chemical reaction (3) of bond separation in S contains 2 types of reagents:


Structures were optimized at the (RI)MP222,23 / cc-pVTZ 24 level of theory using the 5.0.4 version of the ORCA quantum chemistry package. 25 Models were initially prepared using the OpenBabel 26 and Avogadro 27 software. None constraints were applied either in the course of geometry/energy optimization resulted in E2 energy values or in the following finite difference Hessian computations supplied the optimized PES points with the quantum ZPE correction value EZPE. All optimized structures are herewith characterized as PES minima by all eigenvalues of molecular Hessian being positive.
Results and Discussion
First, it should be mentioned that elementary paths of conformational conversion in bicyclo[3.3.1]nonane derivatives drawn here by arrows are hypothetical. Here, they are based upon the known least energy paths of conformational change for 6-membered rings. Each arrow should be assigned to an elementary transformation, characterized by a transition state, but none of them is characterized by now.
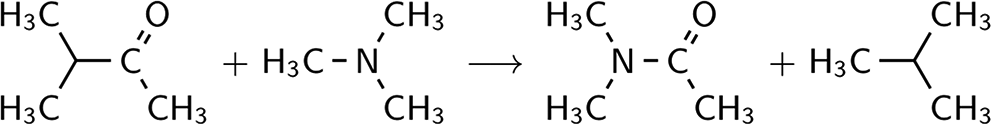
To “turn” an atomic or a bond type into a corresponding reagent molecule is to fill the free valency of the corresponding molecular subgraph—for example, N → −N < or CO → >C=O—by an appropriate univalent “ending” group. The simplest way is their formal “hydrogenaton”: N → NH3, CO → H2CO, N−CO → H2N−CHO. There are no special restrictions to use any other compliant group, other than the context in which the BSR approach is applied. Here, we use “methylation”: N → N(CH3)3, CO → (H3C)2CO, N−CO → (H3C)2N−COCH3 that is shown to be both practical and accurate in the general context of computational organic thermochemistry. The absolute energy values for reagents at this level of theory are published elsewhere for most of molecules to be used hereafter8,21,28 (summarized in the “Supporting Information” section; Table S1 for prototypes, Table S2 for atomic reagents) with a valuable exception of N,N-dimethylacetamide required to estimate the strain in molecules containing the tertiary amide fragment: >N−C(=O)−. From our calculations, we found E2 = −287.26163 au; EZPE = 0.13141 au for this molecule. The energy of “tertiary amide resonance” could be estimated as −17.85 kcal mol−1 through the energy effect of the following reaction:

changing both π and σ-character of the N atom binding environment, and −16.85 kcal mol−1 from the reaction:

where π-conjugation with carbonyl group is introduced. Another related reaction of π-system extension:
gives −18.71 kcal mol−1. The small interval of ±1 kcal mol−1 that contains all these values reveals the small amount of σ-contribution to the amide bond stabilization, the π-effect to be sufficiently greater. It should be mentioned that these values are close to the energy of “primary amide resonance” calculated as −19.6(3) kcal mol−1 from the experimental thermochemistry, and −18.3 kcal mol−1 from high level ab initio calculations. 17 The intricate theoretical question of the amide groups equilibrium (non)planarity is left for further possible special publications.
The general bond separation scheme for 3,7-acetylbispidines (Figure 4) includes both the skeletal bonds and the amide substituent attachments. The equations derived for the investigated structures

The general scheme of bond separation in 3,7-diacetylbispidines. Separated bonds are given in bold.




They all satisfy the criteria of hyperhomodesmotic reaction scheme (atom and bond types being equal in total numbers of corresponding atoms and bonds for both left and right parts of an equation) as extended consistently from that initially formulated for hydrocarbons29,30 to cover heteroatomic molecules, 28 while the amide resonance schemes are homodesmotic. Here, the negative values of Eς exhibit the successful realization of the double “amide resonance” conjugation phenomena as it is mentioned in literature.17,31
All conformers assumed from the scheme of skeleton / substituents conformation coupling in Figure 3 were optimized and characterized as PES minima by Hessian calculations. The results are collected in the “Supporting Information” section (Table S3). Corresponding strain and conformational energy values are presented in Table 1. In all investigated molecules, the “double chair” skeletal forms with antiparallel acetyl groups orientation (
Strain and Conformational Energies of 3,7-Diacetylbispidines

Bispidinones
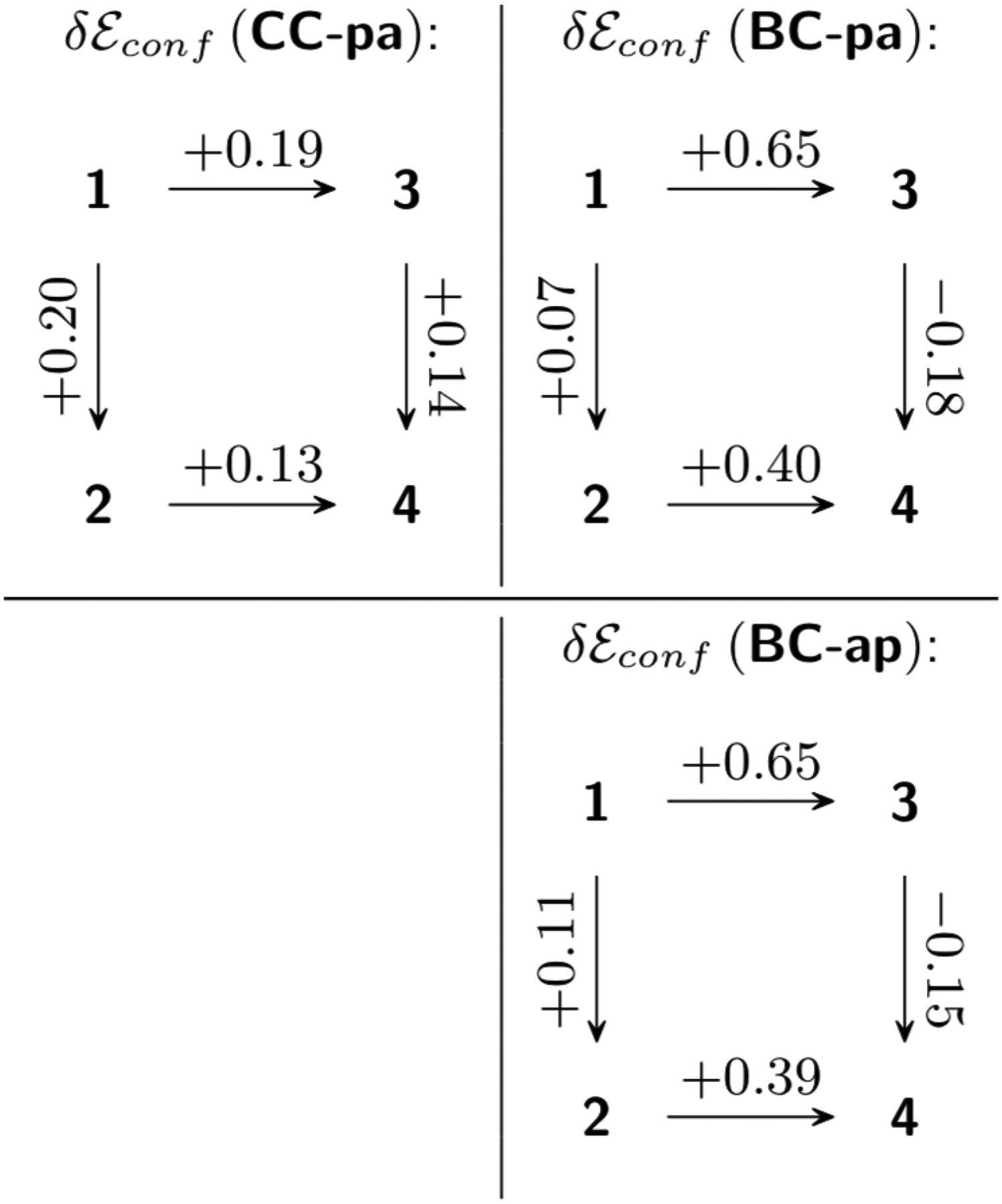
Substitution conformational effects in least-energy conformers, kcal mol–1.
Conclusions
Correlated ab initio calculations on the structures and energy of conformers of 4 bispidine (3,7-diazabicyclo[3.3.1]nonane) derivatives allow to add some points to the known picture of their conformational behavior. Conformations with antiparallel acyl group orientation in 3,7-diacetylbispidines are shown to be lowest in energy and thus dominating the parallel-oriented forms in the equilibrium ensemble. Studies of the conformational behavior show that
Supplemental Material
sj-docx-1-npx-10.1177_1934578X241239199 - Supplemental material for Ab Initio Conformational Analysis of 3,7-Diacetyl-3,7-Diazabicyclo[3.3.1]Nonanes *
Supplemental material, sj-docx-1-npx-10.1177_1934578X241239199 for Ab Initio Conformational Analysis of 3,7-Diacetyl-3,7-Diazabicyclo[3.3.1]Nonanes * by Sergey A. Pisarev, Elena A. Golubeva and Vladimir A. Palyulin in Natural Product Communications
Footnotes
Acknowledgments
The authors thank Russian Science Foundation (grant No. 22-15-00041) for the support of this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Russian Science Foundation, (grant number 22-15-00041).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.